Ru 掺杂对Fe 催化剂上酚类化合物加氢脱氧影响的密度泛函理论研究
2022-08-13聂小娃郭新闻
程 琪, 聂小娃, 郭新闻
(大连理工大学 化工学院 精细化工国家重点实验室, 辽宁 大连 116024, 中国)
1 研究背景
随着经济社会的高速发展, 煤、 石油、 天然气等传统化石能源日益匮乏, 能源紧缺和环境污染的问题逐渐凸显, 因此, 寻找环境友好并可以再生的替代能源迫在眉睫. 生物质作为分布广泛、 储量丰富的可再生资源, 越来越引起人们的关注. 生物质资源来源于光合作用, 合成原料和燃烧后产物都是二氧化碳, 满足绿色、 可持续发展的社会需求. 木质素是生物质的重要组成部分[1], 是芳香性天然高聚物, 具有较大的应用潜力和开发价值[2]. 在木质素转化获得油品的酚类化合物中, 含有大量的酚羟基和与芳环相连的甲氧基[3]. 生物油中酚类的存在使其具有较高的氧含量, 从而造成生物油具有黏性、 腐蚀性、 热值低、 化学稳定性差等不利因素[4],无法作为运输燃料, 不利于大规模应用, 因此必须通过精制来降低氧含量[5], 在不减少碳的同时脱去氧. 催化加氢脱氧(Hydrodeoxygenation, HDO)是目前生物油升级提质的关键步骤, 可以高效地提升生物质油的品质, 同时也符合C 原子和H 原子的经济性. 木质素的热解产物是酚类化合物, 催化加氢脱氧过程可以高选择性地断裂酚类化合物的CAr-O 键, 有效降低热解产物中较高的含氧量, 得到组成成分在C6-C10之间的液态烃类燃料[6], 提高其利用价值. 酚类分子HDO 反应得到的脱氧产物主要是芳烃和环烷烃, 其中芳烃产物的辛烷值高、 附加值高、 耗氢量低, 是化工工业重要的基础原料[7].在催化加氢脱氧反应过程中存在一些副反应, 如芳环加氢饱和、 烷基转移以及C-C 氢解开环等, 这些副反应的发生会消耗更多的H2, 并生成混合产品,因此, 提高酚类加氢脱氧生成芳烃的选择性显得尤为重要. 近年来, 对木质素基酚类化合物加氢脱氧的研究关注点主要在于对催化剂种类的筛选和反应条件的优化, 以期提高对芳烃产物的选择性. 由此可见, 通过优化设计新型催化剂组成和结构, 在相对温和反应条件下提高木质素基酚类化合物加氢脱氧生成目标产物芳烃的反应性能, 具有重要的科学研究价值和实际意义.
酚类加氢脱氧一般分为三种路径: 一是直接脱氧(Direct Deoxygenation, DDO); 二是先加氢再脱氧(Hydrogenation-Deoxygenation, HYD); 三是反应物先异构化生成酮再加氢脱氧(Tautomerization-HYD). DDO 和HYD 路径可以决定芳烃产物与芳环加氢产物的相对选择性, 是主要反应路径. Sun 等[8]的工作表明, 催化剂的类型能够影响愈创木酚是否进行HDO 反应, 并提出了愈创木酚在Pd、 Pt、 Ru、Cu、 Fe 和Pd-Fe 等负载金属催化剂上的反应机理.愈创木酚作为反应物, 一条路径是愈创木酚直接脱氧生成苯酚, 另一条路径是愈创木酚加氢生成邻苯二酚, 再经过直接脱氧生成苯酚. 在苯酚的进一步转化中, 一条路径是先异构化生成酮再加氢脱水,另一条路径是直接脱氧生成目标芳烃产物, 进一步加氢可能生成C1、 C2气相烃类产物. 苯酚先加氢再脱水生成的环己酮、 环己醇等环饱和副产物对产物用作生物油燃料会产生负面影响, 是不希望发生的反应路径. 因此, 如何通过催化剂设计和反应条件优化来控制反应路径从而高选择性地生成芳烃成为研究的重点. 结果表明: 负载金属催化剂催化愈创木酚HDO 反应的主要中间体是苯酚, 与碳负载单金属催化剂相比, Pd-Fe/C 双金属催化剂对HDO 反应生成芳烃具有更高的活性和选择性.
铂系金属(Pt、 Pd、 Ru、 Rh)催化剂凭借其较高的加氢活性被应用于酚类化合物的加氢脱氧反应中. Chang 等[9]研究了Ru/C 和Mo/C 催化愈创木酚加氢脱氧反应的性能, 两种催化剂在4.0 MPa 和380 ℃下均表现出良好的加氢脱氧性能, 产物苯的选择性分别达到了69.5%和83.5%. 两种催化剂的加氢脱氧反应机理有所不同, Ru/C 催化剂上愈创木酚主要反应路径是O-CH3键断裂后形成邻苯二酚,其再进一步加氢脱氧生成苯; Mo/C 催化剂上愈创木酚主要通过C-OCH3键断裂直接脱氧生成芳烃.Bjelic´等[10]考察了温度、 压力等条件对在Ru/C 催化剂上加氢脱氧反应动力学的影响, 结果表明丁香酚中烯丙基和苯环加氢的反应比脱羟基和脱甲氧基的反应更容易发生. 贵金属催化剂上易发生芳环加氢饱和及C-C 氢解开环生成小分子气相产物, 不利于碳原子的有效利用和原子经济性, 同时也限制了芳烃的选择性. 实验研究发现石墨烯负载钌纳米粒子催化剂表现出良好的加氢脱氧性能[11], Rubes等对苯酚在Ru/C 催化剂上的DDO 反应机理进行了DFT 计算, 结果表明, 新的Ru-CAr键形成促进了CAr-O 键活化断裂, 导致苯酚直接加氢脱氧的反应路径明显具有动力学优势, 同时, 氢的存在会导致Ru-O 键更加稳定, 从而进一步促进苯酚通过DDO 路径生成芳烃. 通常载体的加入能够提高催化剂的稳定性和使用寿命, 而且一些特殊载体(如氧化还原性载体)能够对催化活性和产物选择性起到一定调节作用. Newman 等[12]研究了Ru 负载在C、 SiO2、 Al2O3、 TiO2不同载体上催化苯酚加氢脱氧反应. 其中, 高度分散的Ru/TiO2催化剂对苯酚通过DDO 途径加氢脱氧具有最高的活性和选择性, 芳烃的选择性达到86%, Ru 能够促进H2活化和解离, 表面上H 覆盖度的提高有利于CAr-O 键断裂, 从而提高了DDO 反应选择性. Olcese等[13]研究了以SiO2为载体的Fe 金属催化剂上愈创木酚的加氢脱氧反应, 在最佳工艺条件下, 愈创木酚转化为芳烃的转化率为74%, 芳烃(苯、 甲苯、二甲苯, BTX)选择性为38%. 比较单一的金属催化剂和掺杂贵金属的双金属催化剂, 亲氧金属Fe 与贵金属组成的双金属催化剂对愈创木酚和苯酚的加氢脱氧生成芳烃产物表现出较好的选择性. 美国西北太平洋国家实验室Sun 等[8]对Fe、 Pd、 Ru 等单金属催化剂以及Fe-Pd 双金属的加氢脱氧性能进行了实验及计算研究, 结果表明掺杂贵金属后的Pd-Fe 双金属催化剂能够有效增强愈创木酚加氢脱氧的催化活性, 在450 ℃反应温度下, 苯、 甲苯和三甲苯的产率为83.2%.
在酚类加氢脱氧反应过程中, 转化路径和产物分布的调控变得相对复杂, 催化活性和芳烃选择性还有待于进一步提升. 虽然在Fe 基催化剂中引入贵金属对酚类加氢脱氧提高芳烃选择性具有潜在的优势, 但贵金属组分的加入量、 引入方式, 其与Fe活性中心的作用形式, 进而如何影响酚类加氢脱氧反应性能等仍然是需要深入探讨和阐明的重要科学问题. 酚类化合物加氢脱氧, 既要保证一定水平的HDO 活性, 同时还要提高对芳烃的选择性, 抑制芳环加氢饱和及C-C 氢解开环, 降低H2消耗, 反应条件温和, 减少能耗, 这就要求对双金属催化剂的组成结构、 双组分之间以及金属与载体之间的相互作用等进行精准调控, 然而高效催化剂的优化设计仍然面临诸多挑战. 我们采用密度泛函理论计算,在分子水平上对Ru 掺杂Fe 催化剂的表面结构、 电子作用机制、 酚类反应物吸附活化及加氢脱氧反应机理进行了系统研究, 揭示了Ru 掺杂方式及含量对反应路径和反应性能的重要影响, 阐明Ru-Fe 催化剂的潜在构-性关系, 对未来高效加氢脱氧催化剂的设计合成具有一定指导意义.
2 计算方法
2.1 电子结构方法
采用密度泛函理论( Density Functional Theory,DFT ), 它是一种研究多电子体系电子结构的量子力学方法. 使用奥地利维也纳大学Hafner 小组开发的VASP(Vienna Ab initio Simulation Package )软件包进行模拟计算, VASP 软件采用赝势方法和平面波基组展开来处理电子波函数. 交换和相关能采用自旋极化(Spin-polarized)广义梯度近似(GGA)处理, 计算泛函为Perdew Burke Ernzerhof (PBE)[14].平面波基组的截断能为400 eV. 结构优化的收敛标准设置为所有原子上的力常数小于0.2 eV/nm, 能量收敛标准设置为10-5eV. 通过CI-NEB (The climbing image nudged elastic band)方法[15]计算过渡态, 使用包括初态和终态在内的6~8 个图像来搜索过渡态形成的最小能量路径, 并确定每个过渡态沿反应坐标方向有且仅有1 个虚振动频率. 电荷计算采用Henkelman 课题组开发的巴德电荷(Bader charge)计算方法[16].
通过吸附能计算确定稳定吸附构型; 通过计算反应能及活化能垒确定最优反应路径.
表面物种的吸附能(Eads)计算公式为:

其中Eslab-species为吸附物种与表面的整体能量,Eslab为空表面的能量, Especies为吸附物种在气相中的能量. 根据吸附能的定义, 负值越大, 吸附分子与催化剂表面的相互作用越强, 对应的吸附构型就越稳定.
催化剂表面上基元反应的反应能(Erxn)及活化能垒(Eact)计算公式为:

其中FS 为基元反应终态, IS 为反应初态, TS为反应过渡态.
2.2 表面模型
我们在前期工作中对比了Fe 不同晶面对苯酚吸附活化及加氢脱氧性能的影响, 研究结果表明:阶梯状Fe(211)晶面对苯酚的直接脱羟基再加氢生成苯更具催化活性, 有利于目标产物苯的生成; 相比之下, Fe(110)和Fe(111)晶面易于发生苯环加氢反应生成环饱和产物[17]. 因此, 我们选取Fe 的(211)晶面作为催化剂表面模型, 晶格参数采用实验值(0.2866 nm). 体相Fe 是体心立方(bcc)结构,构建的Fe(211)表面采用p(3×3)超胞(晶胞常数为1.489 nm×1.216 nm×1.551 nm), 包 括4 个 原子层. 在结构优化过程中, 位于底部的2 层Fe 原子被固定, 位于顶部的2 层Fe 原子以及吸附分子在优化过程中完全放开. 表面计算中的k点采用(2×2×1), 同时采用( 3×3×1)k点对酚反应物的吸附能进行了测试, 结果表明(2×2×1)k点能够满足计算精度要求(吸附能差异<0.02 eV). 为了避免重复单元之间的相互作用, 真空层的厚度设置为1.2 nm.
为了考察Ru 掺杂方式的影响, 分别采用单Ru取代Fe(211)表面上top 位的1 个Fe 原子(1Rusub-Fe(211))以及单Ru 吸附在Fe(211)表面的hollow 位点(1Ruads-Fe(211))两种方式. 当Ru 原子取代1个表面Fe 原子时, Ru 原子与2 个top 位Fe 原子和2 个hollow 位Fe 原子成键, 高度与top 位Fe 原子平齐, 1Rusub-Fe(211)表面上Ru-Fe 键平均键长为0.251 nm, 比纯Fe(211)表面上Fe-Fe 键平均键长(0.258 nm)短0.007 nm(图1A). 当Ru 原子吸附在hollow 位Fe 原子上时, 其同时与top 位Fe原子成键, 吸附的Ru 原子的位置高于top 位Fe 原子, Ru-Fe 键平均键长0.252 nm(图1B, 此结构为最稳定模型). 为了考察Ru 掺杂量对反应分子吸附以及反应性能的影响, 在单Ru 吸附结构基础上, 构建了4Ru 原子簇吸附在Fe(211)表面上的稳定结构(4Ruads-Fe(211)), 最优结构显示4Ru 原子簇在Fe(211)表面上的分布形状接近铲型, 3 个Ru 原子位于hollow 位Fe 原子上方并与表面top 位和hollow 位Fe 原子成键, 1 个Ru 原子位于bridge位并与hollow 位Fe 原子和Ru 原子成键(图1C),Ru-Ru 键平均键长为0.249 nm, Ru-Fe 键平均键长为0.251 nm.
3 结果与讨论
3.1 不同Ru-Fe(211)表面的电子性质
为了比较Ru 不同引入形式及不同含量对Fe催化剂表面电子性质的影响, 计算了不同Ru-Fe(211)表面上的电荷转移情况. 图1(b)给出了不同Ru-Fe(211)表面的电荷密度图, 结果表明Fe(211)表面上掺杂Ru 原子之后, Fe 原子上的电子向Ru 原子转移. 其中, 1Rusub-Fe(211)表面有0.49 e-的电荷从Fe 表面转移到Ru, 1Ruads-Fe(211)表面Fe 向Ru 原子转移了0.52 e-电荷, 4Ruads-Fe(211)表面Fe 向Ru 簇转移的电荷最多, 为1.33 e-. 比较不同的引入方式, Ru 原子通过吸附的方式引入Fe(211)表面时, Ru 原子与Fe(211)表面的相互作用更强, 电荷转移更多; 比较不同Ru 含量, 4Ru 吸附的Fe(211)表面电荷转移多于1Ru 吸附, 因此Ru-Fe 相互作用增强. 计算结果表明不同引入形式及不同Ru 含量导致Fe 催化剂表面结构及表面电子性质的改变, 进而可能对酚反应物的吸附活化及加氢脱氧反应产生影响.

图1 (A)1Rusub-Fe(211); (B)1Ruads-Fe(211); (C) 4Ruads-Fe(211)表面的(a)稳定构型(b)电荷密度图(蓝色表示电子密度增加, 红色表示电子密度减少)Fig.1 The (a) stable surface configuration and (b)electronic charge density difference of (A) 1Rusub-Fe(211) surface;(B)1Ruads-Fe(211) surface; (C) 4Ruads-Fe(211) surface(Blue and red isosurfaces represent the charge accumulation(i.e., gain of electron density) and depletion (i.e., loss of electron density) in the system, respectively)
3.2 酚类化合物在1Ru-Fe(211)表面上的吸附活化
3.2.1 酚类化合物的吸附性能 Fe(211)表面呈阶梯状, 吸附位点包括六重中空位点(6-fold hollow site)、 顶点(top site)和桥位点(bridge site). 表1比较了同一表面上酚类分子的垂直吸附结构和水平吸附结构的吸附能, 结果表明水平吸附结构吸附能绝对值更大, 酚类分子与金属表面的相互作用更强,因此结构更稳定. 选取了苯酚、 邻甲酚及愈创木酚3种分子作为典型代表, 在考察酚类分子在1Ru-Fe(211)表面上吸附时, 考虑了两种结构: 一种是垂直吸附, 酚类分子-OH基团的O原子与表面上的Fe原子成键(如图2中③); 另一种是水平吸附, 酚类分子苯环与表面发生相互作用并成键(如图2中①). 通过比较吸附能大小找到1Rusub-Fe(211)表面和1Ruads-Fe(211)表面上苯酚、 邻甲酚及愈创木酚的最稳定吸附结构, 分析了电荷转移情况并绘制电荷密度图(如图2中②).

表1 1Rusub-Fe(211)表面和1Ruads-Fe(211)表面上酚类垂直和水平吸附的吸附能(Eads/eV)Table 1 Adsorption energy of vertical and horizontal adsorption of the three phenol reactants on the 1Rusub-Fe(211)and 1Ruads-Fe(211) surface (Eads/eV)
如图2A 中①所示, 在1Rusub-Fe(211)表面上,苯酚、 邻甲酚、 愈创木酚的稳定吸附结构为水平吸附, 苯环与表面趋近平行, 酚类分子的苯环位于阶梯状表面的凹槽中, 由于Fe 表面对H 原子存在排斥作用, 导致酚类分子苯环上的C-H 键向上翘起,苯环上连接的H 原子远离表面, 酚类分子的-OH基团向靠近表面的方向倾斜, 羟基上的H 原子吸附在阶梯状表面的hollow 位点. 苯酚在1Rusub-Fe(211)表面上最稳定吸附结构的吸附能为-1.22 eV, 在最稳定吸附构型中, 4 个苯环上的C 原子与表面上的Fe 原子成键, 平均键长为0.220 nm, 1 个苯环上的C原子与表面上的Ru 原子成键, 键长为0.219 nm; 苯酚和表面之间的电荷转移主要发生在苯环和催化剂表面之间, 共转移0.86 e-电荷. 邻甲酚在1Rusub-Fe(211)表面上最稳定吸附结构的吸附能为-1.05 eV,邻甲酚的甲基位于top 位Fe 原子上方, 朝远离表面的方向翘起, 苯环上的3 个C 原子与表面上的Fe原子成键, 平均键长为0.219 nm, 1 个苯环上的C 原子与表面上的Ru 原子成键, 键长为0.219 nm; 邻甲酚和催化剂表面之间发生0.88 e-电荷转移. 愈创木酚在1Rusub-Fe(211)表面上最稳定吸附结构的吸附能为-1.03 eV, -OCH3基团的O 原子与top 位的Fe原子成键, 键长为0.222 nm, 与O 原子相连的-CH3朝远离表面的方向翘起, 苯环上2 个C 原子与表面上Fe 原子成键, 平均键长为0.215 nm, 1 个C 原子与Ru 原子成键, 键长为0.217 nm; 愈创木酚和表面之间发生0.88 e-电荷转移.

图2 (A)1Ruads-Fe(211)表面和(B) 1Ruads-Fe(211)表面上(a)苯酚(b)邻甲酚(c)愈创木酚的①水平吸附稳定结构及其②电荷密度图和③垂直吸附稳定结构Fig.2 ①Stable adsorption structures; ②electronic charge density difference; ③vertical adsorption structure of phenol, o-cresol and guaiacol on (A) 1Rusub-Fe(211) surface and (B) 1Ruads-Fe(211) surface
如图2B 中①所示, 在1Ruads-Fe(211)表面上,苯酚、 邻甲酚、 愈创木酚最稳定吸附结构为水平吸附, 苯环位于阶梯状表面的凹槽内且与表面接近平行, 苯环上在羟基对位的C原子与Ru原子成键, 苯环上的H原子远离表面翘起, 羟基上的H原子吸附在hollow位点, 羟基向靠近Fe表面的方向倾斜. 苯酚在1Ruads-Fe(211)表面上最稳定吸附结构的吸附能为-1.31 eV, 4个苯环C原子与表面Fe原子成键,平均键长为0.225 nm, 在羟基对位的苯环C原子与Ru原子成键, 键长为0.210 nm; 苯酚与表面之间发生0.97 e-电荷转移. 邻甲酚在1Ruads-Fe(211)表面上最稳定吸附结构的吸附能为-1.06 eV, -CH3基团位于top位Fe原子上方, 向远离表面的方向翘起, 5个苯环C原子与表面Fe原子成键, 平均键长为0.228 nm, 1个C原子与Ru原子成键, 键长为0.209 nm; 分子与表面之间转移0.99 e-电荷. 愈创木酚在1Ruads-Fe(211)表面上最稳定吸附结构的吸附能为-1.14 eV, -OCH3基团的O原子与top位Fe原子成键, 键长为0.221 nm, -CH3朝向远离表面的方向翘起, 苯环上3个C原子与表面Fe原子成键, 平均键长为0.225 nm, 苯环上1个C原子与表面Ru原子成键, 键长为0.214 nm; 分子与表面之间发生0.77 e-电荷转移.
3.2.2 不同酚类化合物的吸附活化性能比较 表2给出了酚类分子在不同1Ru-Fe(211)表面上吸附构型的吸附能和关键结构参数. 在酚类分子的稳定吸附结构中, 苯环上的C原子与表面上的Fe原子和Ru原子成键, 分析了Fe-C键平均键长以及Ru-C键键长与吸附能之间的关系: 在1Rusub-Fe(211)表面上, 吸附能从大到小依次为苯酚、 邻甲酚、 愈创木酚, 苯酚稳定吸附结构的Fe-C键平均键长和Ru-C键键长最大, 其次是邻甲酚, 键长最短的是愈创木酚; 如图3所示, 酚类分子在1Rusub-Fe(211)表面上吸附时, Fe-C键平均键长和Ru-C键键长越大, 吸附能更大, 吸附结构更稳定. 在1Ruads-Fe(211)表面上, 苯酚的吸附能最大, 其次是愈创木酚, 邻甲酚的吸附能最小, 邻甲酚稳定吸附结构的Ru-C键键长最小, Fe-C键平均键长最大. 比较3种酚在不同1Ru-Fe(211)表面上的吸附活化参数, 发现在1Ruads-Fe(211)表面上的吸附结构与其在1Rusub-Fe(211)表面上的吸附结构相比, Fe-C键平均键长更长, Ru-C键键长更短, 酚分子与催化剂表面之间转移电荷更多、 相互作用更强, 吸附能更大, 吸附结构更稳定. 因此, 1Ruads-Fe(211)表面更有利于酚分子的吸附活化.

表2 苯酚、 邻甲酚、 愈创木酚在1Rusub-Fe(211)和1Ruads-Fe(211)表面上的吸附能和关键结构参数Table 2 Adsorption energy and key structural parameters of phenol, o-cresol and guaiacol on the 1Rusub-Fe(211) and the 1Ruads-Fe(211) surface
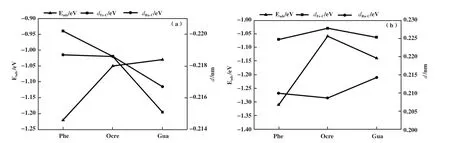
图3 (a) 1Rusub-Fe(211)及(b) 1Ruads-Fe(211)表面上3种酚的吸附能与Fe-C键长及Ru-C键长之间的关系Fig.3 Relationship between the adsorption energy and Fe-C bond length and Ru-C bond length of the three phenol reactants on(a)1Rusub-Fe (211) surface and (b) 1Ruads-Fe (211) surface
3.2.3 H*表面覆盖度对酚类化合物吸附稳定性的影响 在不同1Ru-Fe(211)表面上计算了H2的吸附、活化和解离过程. 在1Rusub-Fe(211)表面上, H2分子在Ru原子上可以发生直接解离, 解离后的H原子吸附在top位Fe原子和top位Ru原子之间的bridge吸附位点, 解离吸附能为-1.29 eV(取代表面上H2吸附结构优化后直接解离为2个H原子吸附; 此处解离吸附能为2个H原子在取代表面上稳定吸附结构的吸附能). 在1Ruads-Fe(211)表面上, H2分子的吸附能为-0.50 eV, H2分子解离后的H原子分别吸附在hollow位和bridge位, 解离能垒为0.12 eV, 过渡态的H-H距离为0.121 nm, 解离吸附能为-1.17 eV(吸附表面上H2吸附结构优化后未发生解离, 构建解离后的FS并优化; 此处解离吸附能为FS结构的吸附能, 即: FS能量-气相中H2能量-空表面能量).根据文献数据, H2分子在纯Fe(211)表面上解离吸附能为-1.14 eV, 在1Pd-Fe(211)表面上解离吸附能为-1.25 eV[18], 对比表明, 在Fe(211)表面添加Ru能够促进H2的活化和解离, 为加氢脱氧反应提供更多的活性H*物种, 提高反应速率. 当表面H*覆盖度为1H*和3H*时, 3种酚反应物的吸附结构与0H*时的吸附结构相比没有明显变化, 因此, H*物种表面覆盖度对酚的吸附构型影响不大. 表3给出了不同H*覆盖度下苯酚、 邻甲酚、 愈创木酚在两种1Ru-Fe(211)表面上的吸附能, 可以看出, H*物种表面覆盖度对酚的吸附强度影响不明显, 吸附能变化<0.05 eV. 因此在研究加氢脱氧反应机理时, 没有考虑H*表面覆盖度的影响.
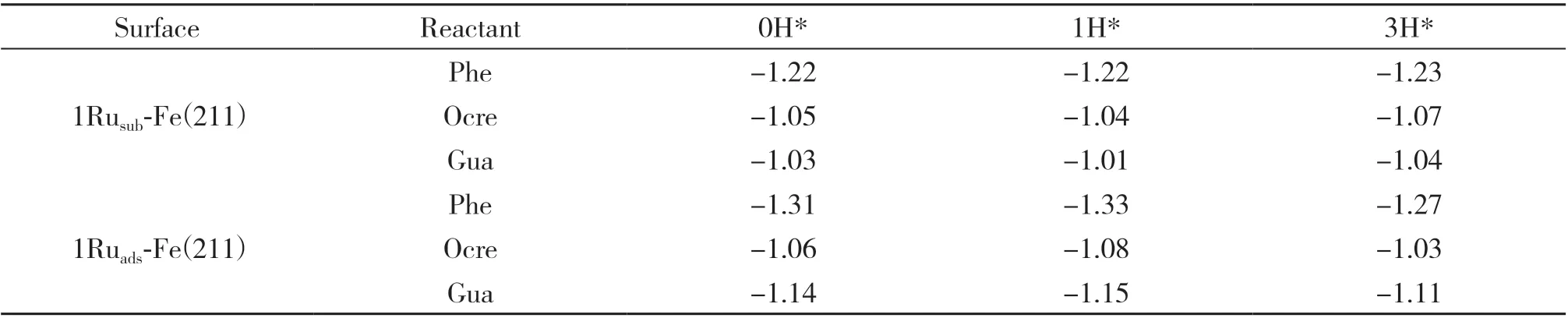
表3 不同H*覆盖度的1Rusub-Fe(211)和1Ruads-Fe(211)表面上苯酚、 邻甲酚、 愈创木酚的吸附能(Eads/eV)Table 3 Adsorption energy of phenol, o-cresol and guaiacol on 1Rusub-Fe(211) and 1Ruads-Fe(211)surfaces with different surface H* coverage (Eads/eV )
3.3 酚类化合物在1Ru-Fe(211)表面上的加氢脱氧
3.3.1 酚类化合物直接加氢脱氧反应机理 以苯酚、 邻甲酚、 愈创木酚最稳定吸附结构为基础, 分别在两种1Ru-Fe(211)表面上计算了酚类化合物加氢脱氧反应的过渡态和反应路径. 其中, 苯酚脱羟基后再加氢的产物是苯; 邻甲酚脱羟基后再加氢的产物是甲苯; 愈创木酚加氢脱氧生成苯有3种路径:一种是脱-OH后再加氢生成苯甲醚, 苯甲醚脱-CH3后再加氢生成苯酚, 苯酚再继续转化生成苯; 一种是脱-CH3后生成邻苯二酚, 其再脱2个-OH后生成苯; 一种是脱-OCH3后再加氢直接生成目标产物苯.3.3.2 1Rusub-Fe(211)表面上的反应路径 如图4A所示, 在1Rusub-Fe(211)表面上, 苯酚脱羟基时其CAr-O键断裂, 在表面上bridge位点形成吸附态的OH*物种, OH*物种的H原子朝向远离表面的方向,C6H5*中间体中原本与-OH相连的CAr原子距离金属表面更近, 并与top位Fe原子成键. 苯酚脱羟基过渡态的CAr-O键距离为0.198 nm, 反应能垒为0.86 eV,反应能为-0.91 eV; 苯酚脱羟基后形成的C6H5*中间体继续加氢, 能垒为0.52 eV, 反应能为-0.06 eV. 如图4B所示, 邻甲酚脱羟基时其CAr-O键断裂, 形成的C7H7*中间体的CAr原子靠近金属表面且与top位Fe原子成键, -CH3朝向远离表面的方向, 邻甲酚脱羟基过渡态的CAr-O键距离为0.194 nm, 反应能垒为1.14 eV, 反应放热0.65 eV; 邻甲酚脱羟基后形成的C7H7*中间体继续加氢需要克服0.44 eV能垒, 反应能为-0.48 eV. 如图4C(a)所示, 在愈创木酚直接脱甲氧基的反应中, 脱甲氧基能垒为1.16 eV, 反应放热0.99 eV, 中间体C6H5O*继续加氢能垒为0.69 eV,反应放热0.16 eV, 脱甲氧基步骤为控速步骤, 直接加氢脱氧的有效能垒为1.16 eV. 如图4C(b)所示, 在愈创木酚脱羟基的反应中, 其CAr-O键断裂后CAr原子与表面top位Fe原子成键, 苯环上的甲氧基和OH*吸附物种均朝向远离表面的方向翘起, 与苯酚和邻甲酚脱羟基过渡态相比, 愈创木酚过渡态的CAr-O键距离更大、 能垒更高, CAr-O键距离为0.211 nm, 反应能垒为1.40 eV, 反应能为-0.69 eV; 愈创木酚脱羟基后再加氢步骤的能垒为0.50 eV, 反应能为-0.48 eV. 愈创木酚脱羟基再加氢的产物苯甲醚继续脱-CH3, C-O键断裂后在表面bridge位上形成-CH3*吸附物种, 反应能垒为0.95 eV, 反应能为-1.39 eV; 苯甲醚脱甲基后形成的C6H5O*中间体再加氢需克服1.28 eV的能垒, 反应吸热1.05 eV. 根据前面的计算结果, 苯酚加氢脱氧生成苯的能垒为0.86 eV, 因此, 在愈创木酚先生成苯甲醚再生成苯的反应路径中能垒最高的是第一步脱羟基(CAr-O键断裂能垒1.40 eV). 如图4C(c)所示, 愈创木酚直接脱甲基的反应能垒为1.25 eV, 反应放热1.45 eV, 形成的C6H5O2*中间体再加氢过程需要克服1.61 eV能垒, 反应吸热1.05 eV, 加氢过程是速控步骤, 因此愈创木酚先脱甲基生成邻苯二酚的路径在动力学上是不利的, 不是芳烃生成的反应路径. 表4 给出了1Rusub-Fe(211)表面上愈创木酚加氢脱氧不同路径的反应能、 活化能及过渡态结构参数, 对比愈创木酚加氢脱氧不同反应路径的能线图(如图5(a)所示), 表明在1Rusub-Fe(211)表面上, 愈创木酚脱甲氧基直接生成苯酚, 再由苯酚加氢脱氧生成苯的路径的反应能垒最低, 反应更容易进行, 因此是芳烃生成的动力学优势路径.
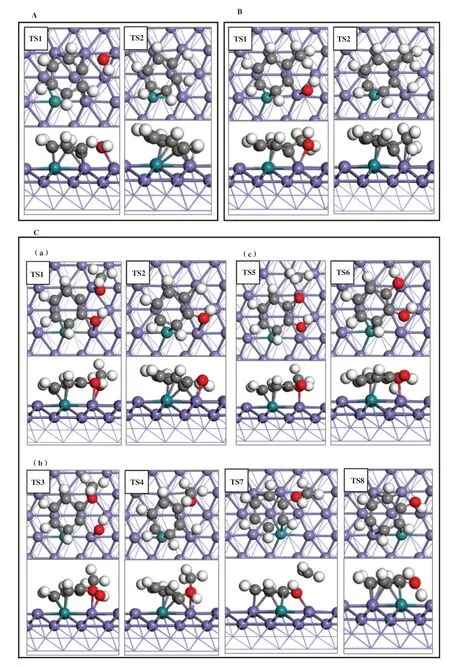
图4 在1Rusub-Fe(211)表面上(A)苯酚脱羟基再加氢; (B)邻甲酚脱羟基再加氢; (C)愈创木酚(a)脱甲氧基直接生成苯酚(b)脱羟基经苯甲醚再生成苯酚(c)脱甲基生成邻苯二酚各过程的过渡态结构图Fig.4 Transition state structures of (A)phenol dehydroxylation followed by hydrogenation (B)o-cresol dehydroxylation followed by hydrogenation (C) guaiacol transformation to(a) phenol from anisole; (b) phenol directly via demethoxylation; (c) catechol through demethylation on the surface of 1Rusub-Fe(211)
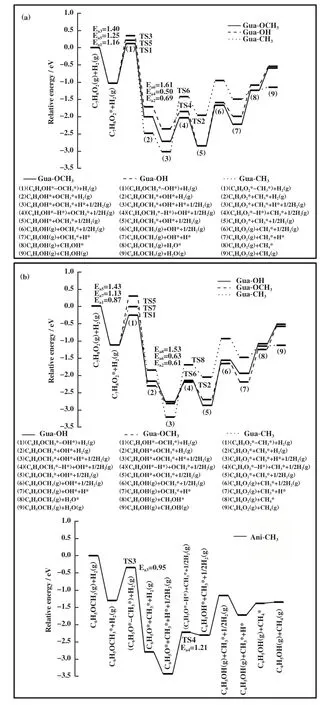
图5 在(a)1Rusub-Fe(211)和(b)1Ruads-Fe(211)表面上愈创木酚加氢脱氧不同路径的反应能线图Fig.5 Energy diagrams of different paths of guaiacol hydrodeoxygenation on the (a)1Rusub-Fe(211) surface and (b)1Ruads-Fe(211) surface
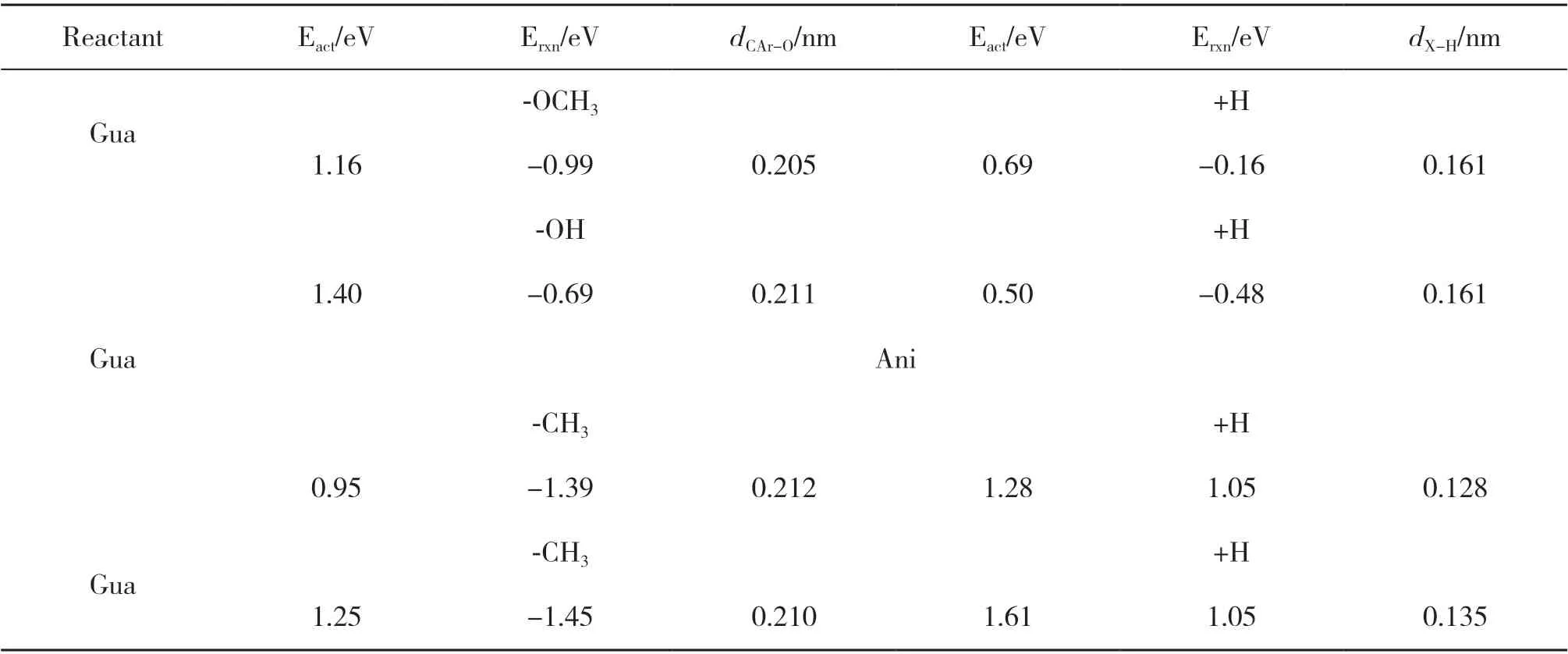
表4 在1Rusub-Fe(211)表面上愈创木酚加氢脱氧不同路径的反应能、 活化能及过渡态结构参数Table 4 Reaction energy, activation barrier and transition state structural parameters of guaiacol hydrodeoxygenation on the 1Rusub-Fe(211) surface
3.3.3 1Ruads-Fe(211)表 面 上 的 反 应 路 径 在1Ruads-Fe(211)表面上, 如图6A所示, 苯酚脱羟基后苯环倾斜角度变大, 原本与Ru原子相连且位于羟基对位的C原子不再与Ru原子成键, CAr-O键断裂后形成的-OH基团吸附在Fe表面上的bridge位点上,OH*物种的H原子远离表面, 过渡态CAr-O键距离为0.183 nm, 反应能垒为0.73 eV, 反应能为-1.09 eV;苯酚脱羟基后加氢的反应能垒为0.49 eV, 反应能为0.02 eV. 如图6B所示, 邻甲酚脱羟基时CAr-O键活化断裂, 在top位Fe原子之间的bridge位点上形成吸附态的OH*吸附物种, 与甲基相连的C原子与表面Fe原子的距离更近, 甲基朝远离表面的方向翘起,过渡态结构的CAr-O距离为0.188 nm, 反应能垒为0.86 eV, 反应放热0.98 eV; 反应生成的C7H7*中间体再加氢过程需克服0.56 eV能垒, 反应能为0.02 eV(如表5所示).
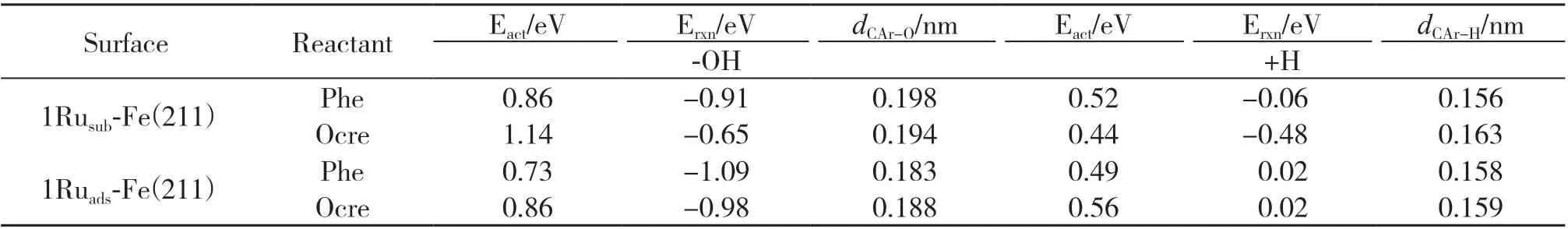
表5 在1Rusub-Fe(211)和1Ruads-Fe(211)表面上苯酚及邻甲酚脱羟基再加氢反应能、 活化能及过渡态结构参数Table 5 Reaction energy, activation barrier and transition state structural parameters of dehydroxylation and hydrogenation of phenol and o-cresol on 1Rusub-Fe(211) and 1Ruads-Fe(211) surfaces

图6 在1Ruads-Fe(211)表面上(A)苯酚脱羟基再加氢; (B)邻甲酚脱羟基再加氢; (C)愈创木酚(a)经苯甲醚生成苯酚(b)直接生成苯酚(c)脱甲基生成邻苯二酚各过程的过渡态结构图Fig.6 Transition state structures of (A)phenol dehydroxylation followed by hydrogenation (B)o-cresoldehydroxylation followed by hydrogenation (C) guaiacol transformation to(a) phenol from anisole; (b) phenol directly via demethoxylation; (c) catechol through demethylation on the surface of 1Ruads-Fe(211)
愈创木酚在1Ruads-Fe(211)表面上脱羟基生成苯甲醚的反应中(如图6C(a)所示), 脱羟基步骤为控速步骤, 能垒为0.87 eV, 反应能为-1.04 eV;苯甲醚脱甲基生成苯酚的反应中, 中间体C6H5O*加氢步骤为控速步骤, 能垒为1.21 eV, 反应能为1.13 eV. 由于苯酚在1Ruads-Fe(211)表面上加氢脱氧最高能垒为0.73 eV, 低于苯甲醚加氢脱氧能垒, 因此愈创木酚通过苯甲醚中间产物生成苯的总反应有效能垒为1.21 eV. 在愈创木酚直接脱甲氧基生成苯酚的反应路径中(如图6C(b)所示), 脱甲氧基后苯环原来与甲氧基相连的C原子与表面上的Fe原子成键,苯环距离表面更近, 反应能垒为1.43 eV, 反应放热1.18 eV; 随后再加氢步骤的反应能垒为0.63 eV, 反应能为0.11 eV, 因此直接脱甲氧基是该路径的速控步骤. 在愈创木酚直接脱甲基通过邻苯二酚中间产物进行加氢脱氧的反应路径中(如图6C(c)所示), 脱甲基反应能垒为1.13 eV, 反应能为-0.74 eV, 而中间体C6H5O2*加氢步骤的反应能垒为1.53 eV, 反应吸热1.14 eV, 因此该反应路径在动力学上不利的, 不是芳烃产物的生成路径. 表6给出了愈创木酚在1Ruads-Fe(211)表面上不同路径加氢脱氧过程的反应能垒及相关参数, 比较愈创木酚加氢脱氧不同反应路径的能线图(如图5(b)所示), 表明在1Ruads-Fe(211)表面上,愈创木酚先脱羟基生成苯甲醚, 苯甲醚继续脱甲基生成苯酚再进一步加氢脱氧生成苯的反应路径更容易进行.

表6 在1Ruads-Fe(211)表面上愈创木酚加氢脱氧不同路径的反应能、 活化能及过渡态结构参数Table 6 Reaction energy, activation barrier and transition state structural parameters of guaiacol hydrodeoxygenation on the 1Ruads-Fe(211) surface
3.3.4 影响酚反应物CAr-OH键断裂性能的因素 如图7所示, 对比不同1Ru-Fe(211)表面上酚类脱羟基的反应能线图发现, 脱羟基步骤的能垒均高于加氢步骤的能垒, 脱羟基步骤为控速步骤; 对比两个表面, 苯酚在1Ruads-Fe(211)表面上CAr-OH键断裂步骤的能垒比在1Rusub-Fe(211)表面上低0.13 eV, 邻甲酚低0.28 eV, 愈创木酚低0.53 eV. 也就是说, 1Ruads-Fe(211)表面更有利于酚类CAr-OH键断裂、 发生脱羟基反应. 在酚类脱羟基的反应过程中,比较反应能垒与过渡态结构的关键参数发现, 在反应能垒更大的过渡态中, CAr-O键距离往往更大,更难活化; 在1Ruads-Fe(211)表面上, 酚类脱羟基反应能垒较大时, 过渡态O-Fe键长更大, -OH基团与表面距离更远. 因此过渡态的结构参数与其稳定性相关, 进而影响CAr-O键断裂活性.

图7 (a) 苯酚; (b)邻甲酚; (c)愈创木酚在1Rusub-Fe(211)表面和1Ruads-Fe(211)表面上脱羟基再加氢过程的反应能线图Fig.7 Reaction energy diagrams of (a) phenol; (b) o-cresol; (c) guaiacol dehydroxylation followed by hydrogenation on 1Rusub-Fe(211) and 1Ruads-Fe(211) surfaces
3.4 Ru-Fe表面结构对酚类吸附和加氢脱氧的影响
3.4.1 酚类化合物在4Ru-Fe(211)表面上的吸附活化 对比酚类在4Ruads-Fe(211)表面上不同吸附位点的吸附构型和吸附能发现, 当吸附分子只与Ru原子成键时, 吸附能更大, 吸附结构更稳定; 当吸附分子同时与Fe原子和Ru原子成键时吸附性能次之;当吸附分子远离Ru原子, 只与表面上Fe原子成键时, 吸附能较小, 吸附结构不稳定. 以苯酚为例, 当苯酚只与Ru原子成键时吸附能为-1.83 eV; 与Fe原子和Ru原子同时成键时吸附能降低为-1.43 eV; 当苯酚远离Ru原子, 只与Fe原子成键时吸附能降低到-1.10 eV, 低于苯酚在1Ruads-Fe(211)表面上的吸附能(-1.31 eV). 因此, Ru-C键键长与酚类化合物的吸附活化性能可能具有一定相关性.
在4Ruads-Fe(211)表面上, 苯酚、 邻甲酚、 愈创木酚最稳定吸附结构为水平吸附, 酚类分子的苯环吸附在Fe(211)表面凹槽处的Ru 原子上方, 大π键与4Ru 原子簇发生相互作用, 苯环上的C 原子与Ru 原子成键, 羟基位于hollow 吸附位点. 其中, 苯酚羟基上的H原子朝向表面倾斜, 吸附能为-1.83 eV,分子与表面之间转移电荷0.49 e-, 在3种酚中吸附能最大, 转移电荷最多; 邻甲酚羟基上的H原子向表面倾斜程度较小, 甲基向远离表面的方向翘起,吸附能为-1.74 eV, 转移电荷0.47 e-; 愈创木酚羟基上的H原子和甲氧基均朝向远离表面的方向翘起, 吸附能为-1.73 eV, 转移电荷0.44 e-, 在3种酚中吸附能最小、 转移电荷最少. 对比1Ruads-Fe(211)和4Ruads-Fe(211)表面, 苯酚、 邻甲酚、 愈创木酚在4Ru-Fe(211)表面上的吸附能更大, 吸附结构更稳定,酚类分子与表面成键的平均键长更短、 转移电荷更少, 吸附分子与表面的距离更近.
3.4.2 酚类化合物在4Ruads-Fe(211)表面上的加氢脱氧 选取苯酚为酚类化合物代表, 计算了苯酚在4Ruads-Fe(211)表面上的加氢脱氧反应性能. 苯酚脱羟基时其CAr-O键断裂, 原来与羟基相连的C原子与Ru原子成键, 形成的OH*物种吸附在表面top位Fe原子之间的bridge位点, 过渡态CAr-O键距离为0.192 nm, 脱羟基过程的反应能垒为1.48 eV, 反应放热0.53 eV; 形成的C6H5*中间体再加氢, 能垒为0.36 eV, 反应能为-0.80 eV. 相比于1Ruads-Fe(211)表面, 苯酚在4Ruads-Fe(211)表面上加氢脱氧的有效能垒提高了0.75 eV, 过渡态CAr-O键距离增大了0.009 nm, CAr-O键更难断裂. 1Ruads-Fe(211)表面比4Ruads-Fe(211)表面更有利于苯酚加氢脱氧反应进行生成芳烃. 对苯酚在两个表面上脱羟基过程形成过渡态结构的电荷进行了分析(如表7所示), 发现4Ruads-Fe(211)表面上形成过渡态结构中, 苯酚与表面之间电荷转移为0.87 e-, 比1Ruads-Fe(211)表面上电荷转移少0.53 e-, 因此形成的过渡态稳定性低于1Ruads-Fe(211)表面, 反应能垒升高.

表7 苯酚、 邻甲酚、 愈创木酚在1Ruads-Fe(211)表面与4Ruads-Fe(211) 表面上吸附活化、 脱羟基反应参数比较Table 7 Comparison of adsorption, activation, and dehydroxylation reaction parameters of phenol, o-cresol and guaiacol on 1Ruads-Fe(211) and 4Ruads-Fe(211) surfaces
上述计算结果表明, 随着Ru 原子数目增加,酚在Ru-Fe 表面上的吸附显著增强, 不利于其发生进一步转化; Fe 向Ru 转移的电荷增加, 而酚反应物的活化减弱, CAr-O 键断裂形成的过渡态稳定性减弱, 导致脱羟基能垒明显升高. 在Fe 催化剂中掺杂Ru 虽然能够促进H2的活化和解离, 但对酚类加氢脱氧反应生成芳烃具有一定影响, 只有当加入少量Ru 时, 才可能有利于CAr-O 键断裂. 此外,Ru 的掺杂形式(表面取代与表面吸附)对酚反应物的加氢脱氧反应路径也具有一定影响, 在1Rusub-Fe(211)和1Ruads-Fe(211)两种表面上, 虽然愈创木酚加氢脱氧生成产物苯的过程都经过苯酚中间产物, 但反应路径却明显不同: 1Rusub-Fe(211)表面上愈创木酚易于发生直接脱甲氧基生成苯酚, 而1Ruads-Fe(211)表面上愈创木酚则先脱羟基生成苯甲醚, 苯甲醚再继续脱甲基生成苯酚. 计算结果表明, Ru-Fe 催化剂表面结构及Ru 的掺杂量对酚类加氢脱氧反应路径和反应性能具有重要影响.
4 结论
通过DFT 计算, 比较了在贵金属表面取代和表面吸附两种引入方式构建的Ru-Fe(211)表面上,苯酚、 邻甲酚、 愈创木酚的吸附活化和加氢脱氧反应路径, 研究了关键参数对反应性能的影响, 比较了生成芳烃产物的优势反应路径, 得到主要结论如下:
(1) 酚、 邻甲酚、 愈创木酚在1Ru-Fe(211)表面上的吸附结构中, 苯环与表面相互作用发生吸附的水平吸附结构优于通过-OH基团进行吸附的垂直吸附结构; 酚反应物在1Ruads-Fe(211)表面上的吸附比在1Rusub-Fe(211)表面上的吸附更稳定. 稳定吸附结构中苯环上的C 原子均与表面上的Ru 原子和Fe 原子成键, 吸附能与Ru-C 键长、 Fe-C 键平均键长等关键结构参数有关. 掺杂金属Ru 有利于H2的快速活化和解离, 导致表面H*物种的覆盖度增加, 提高了加氢脱氧反应速率, 但对酚反应物分子的吸附构型和吸附稳定性影响较小.
(2)酚类分子发生脱羟基再加氢反应的难易程度依次为: 苯酚<邻甲酚<愈创木酚, 与1Rusub-Fe(211)表面相比, 酚类在1Ruads-Fe(211)表面上脱羟基再加氢的反应能垒更低, 过渡态的CAr-O 键距离更小. 不同表面上愈创木酚加氢脱氧生成芳烃的优势反应路径不同: 1Rusub-Fe(211)表面上, 愈创木酚脱甲氧基直接生成苯酚、 苯酚再经加氢脱氧生成产物苯的路径更容易进行; 1Ruads-Fe(211)表面上,愈创木酚先生成苯甲醚再生成苯酚、 苯酚再经加氢脱氧生成产物苯的路径则更具有动力学优势. 因此,贵金属Ru 的引入方式及其对Fe 催化剂表面结构和电子性质的改变能够影响芳烃产物的生成路径.
(3)1Ruads-Fe(211)相比, 4Ruads-Fe(211)表面上3 种酚的吸附更稳定, 酚分子的苯环与表面上的Ru 原子簇相互作用而发生吸附, 苯环与表面的距离更近, 但是酚反应物脱羟基再加氢的反应能垒升高, 因此不利于加氢脱氧反应进行.
理论计算结果表明, Ru 掺杂Fe 催化剂在酚类化合物加氢脱氧反应制备芳烃方面具有潜在优势,并且其掺杂方式和掺杂量对酚类分子的吸附活化和加氢脱氧性能具有重要影响, 为未来高效加氢脱氧双金属催化剂的优化设计提供了理论基础和方向性指导.
