miRNA 在心房颤动发病机制中的作用
2022-07-27刘弟世闻赵庆彦
刘弟世闻 赵庆彦
武汉大学人民医院心内科/武汉大学心血管病研究所/心血管病湖北重点实验室 湖北 武汉 430060
心房颤动(房颤;atrial fibrillation,AF)的机制复杂,与心房和心室心肌结构和电重构密切有关。AF的电生理机制包括:①触发活动(早后除极)引起的局灶性放电;②动作电位时程(action potential duration,APD)缩短导致的折返;③心房纤维化引起冲动传导的异质性。心房纤维化的发生和发展是AF结构重塑的重要标志,是AF 向持续性方向发展的基础,并降低抗心律失常药物治疗的有效性。心肌纤维组织的逐步积累是心肌重塑的重要原因。纤维结缔组织的形成和再分布调节心房的几何形状,以适应病理性应力、化学和电刺激的影响。这种适应过程涉及心肌的细胞成分和细胞外基质的改变。电重构和纤维化相互影响,共同维持着AF 的发生和发展。miRNA 通过作用于靶基因在转录后水平进行调控,近二十年来关于miRNA 对AF 纤维化和电重构的研究已经有了长足的进展。
1 心肌细胞与心肌成纤维细胞的通讯
心肌成纤维细胞在细胞外基质的形成中起关键作用,它占心肌组织的60%。成纤维细胞本质上是不可兴奋细胞,但可以通过纤维连接蛋白在心肌细胞之间传导电流。这种作用可能导致电流传导的异质性,缩短心肌细胞APD,使静息心肌细胞自动去极等。肌成纤维细胞源于心肌成纤维细胞,但合成胶原蛋白的能力是后者的2 倍,并且对促炎和促纤维化刺激反应更强,能够合成多种细胞因子和趋化因子。肌成纤维细胞含有α-平滑肌肌动蛋白和黏附复合物,后者将肌成纤维细胞内部微丝与细胞外基质蛋白结合,从而为细胞外基质提供收缩力。一系列生长因子、细胞因子、激素、机械性应力和缺氧等,调节着细胞外基质和心肌成纤维细胞活性。这些因素决定了成纤维细胞分化、基因表达以及胶原蛋白的合成,对心肌重塑起着重要作用。
心肌细胞和心肌(肌)成纤维细胞之间存在紧密联系,它们通过自分泌和旁分泌的方式进行信号转导。心肌细胞和心肌(肌)成纤维细胞之间有许多共同的信号通路,如血管紧张素(Ang)Ⅱ、转化生长因子β1(TGF-β)、内皮素(ET)等。然而,它们对信号分子的响应程度依据细胞类型而定,成纤维细胞通过AngⅡ诱导释放TGF-β 和ET-1 来调控心肌细胞肥大和凋亡,而心肌细胞通过AngⅡ诱导释放TGF-β 和ET-1 刺激成纤维细胞增殖、分化和细胞外基质的形成[1]。敲除AngⅡ受体基因的成纤维细胞增殖不明显[2]。心肌细胞和心肌成纤维细胞之间在受体密度和受体激酶活性也存在差异,这可能会干扰效应分子的最终作用。如成纤维细胞AngⅡ受体密度更高[2]。除此之外,一些刺激因子主要对成纤维细胞起作用,如血小板源性生长因子(PDGF)、成纤维细胞生长因子2 等。
2 促纤维化信号通路
2.1 TGF-β1 和Ang Ⅱ在众多的调节因子中,AngⅡ和TGF-β1是心肌成纤维细胞分泌胶原蛋白最为有效的刺激物。在细胞外,TGF-β1通过结合二聚化的受体而发挥作用。配体-受体结合导致磷酸化级联反应,最终导致无活性的Smad2/3/4 形成Smad 复合体。Smad 复合体易位至靶细胞核,参与纤维化基因的表达。
Ang 是一种引起血管收缩和血压升高的寡肽。研究表明,AngⅡ和TGF-β1并非彼此独立发挥作用,而是作为心肌重构和纤维化的整合信号通路的一部分。AngⅡ通过心肌细胞和成纤维细胞中的1型Ang 受体上调TGF-β1表达,进一步激活ERK 诱导心脏成纤维细胞中的胶原蛋白形成[3]。在缺乏TGF-β 的情况下,AngⅡ不能在体内诱导心肌肥大和 纤 维 化[4],但 它 可 以 上 调TGF-β1合 成,促 进Smad2 磷酸化和Smad 复合物的核转位并且增加Smad 复合体与DNA 结合。而TGF-β1可以直接刺激Ang 受 体 的 表 达[5]。这 些 结 果 表 明Ang Ⅱ和TGF-β 在体内共同诱导纤维化形成。
2.2 结缔组织生长因子(connective tissue growth factor, CTGF)CTGF 是促进血管生成的细胞基质蛋白,它能促进细胞黏附并增强细胞外配体的黏附。CTGF 可由TGF-β、AngⅡ和ET-1 诱导产生,并且可能是这些蛋白的下游信号通路。CTGF 不是TGF-β 发挥作用所必需,但它可以作为TGF-β的辅因子来诱导纤维化[6]。但就其本身而言,CTGF 促纤维化能力较弱,它更可能是创造一个有利于纤维化的环境。在健康的心肌中,CTGF 主要在成纤维细胞中表达,但是,在心肌重塑过程中,心肌细胞也会分泌CTGF。
2.3 PDGFPDGF 包 含α 和β 两 种 不 同 的 受 体。体外实验证明PGDF 可以促进成纤维细胞向成肌成纤维细胞转化[7]。PDGF-β 受体抑制剂甲磺酸伊马替尼能延迟伤口愈合,并减少肌成纤维细胞数量、纤连蛋白和Ⅰ型胶原蛋白的表达[8]。在博来霉素诱导皮肤纤维化的小鼠模型中,达沙替尼和尼洛替尼能以剂量依赖性的方式降低皮肤厚度、肌成纤维细胞数量和皮肤胶原蛋白含量[9]。通过腺病毒将PDGF 基因转染至小鼠心脏,能显著上调TGF-β1并加速心脏纤维化和动脉硬化,表明PDGF 可以通过升高TGF-β 水平来促进纤维化[10]。与此相反的是,注射PDGF-α 受体特异性抗体可减轻心房纤维化[11]。
2.4 ET-1ET 是一种强大的血管收缩剂,具有促有丝分裂特性,ET 有3 种亚型,其中,ET-1 是人类重要的亚型。TGF-β 可以通过JNK 诱导ET-1,表明ET-1 是成纤维细胞中TGF-β 诱导纤维化下游信号通路产物[12]。AngⅡ也可以通过激活ERK 和活性氧(ROS)的方式诱导ET-1 产生[13]。而ET-1 可以促进细胞外基质形成和成纤维细胞的分化[14]。这些研究表明,ET 是TGF-β/AngⅡ下游信号通路。
2.5 基质金属蛋白酶(MMP)和金属蛋白酶组织抑制剂(TIMP)细胞外基质的重塑不仅包括合成,还包括协同降解。心肌细胞和心肌成纤维细胞合成的MMP 及TIMP 与细胞外基质稳态维持密切相关。MMP1 的过表达早期可引起心肌代偿性肥厚和胶原蛋白沉积,随着时间延长,细胞外基质的过度降解会引起心肌舒张和收缩功能障碍[15]。此外,MMP1 作用产生的胶原蛋白和基质片段本身会形成生物活性分子,并促进细胞外基质炎性因子和纤维化因子释放。这些炎症因子能促进成纤维细胞活化为肌成纤维细胞,通过作为配体刺激结缔组织合成白细胞整合素和激活其他细胞受体。尽管MMP 主要功能是针对基质降解,但其活性产物也解释了MMP 活性高时纤维化进展快。MMP 活性主要由TIMP 和富含半胱氨酸的Kazal 回文序列的蛋白来调节[16]。
2.6 氧化应激和炎症较低水平的氧化应激就能诱导细胞产生超微结构变化,导致细胞结构和功能受损。 活性氧簇(ROS)可来源于线粒体、NAD(P)H、NADPH 氧化酶(NOX)、黄嘌呤氧化酶和未偶联的一氧化氮合酶。NOX 在心肌氧化还原信号转导中占重要地位,并在多种细胞中表达,包括心肌细胞、成纤维细胞、内皮细胞和炎性细胞等。线粒体中ROS 的长期增加会导致线粒体DNA 损伤和细胞凋亡。此外,ROS 能激活多种肥大信号激酶和转录因子。它还可以刺激成纤维细胞增殖并激活MMP,从而导致细胞外基质重塑[17]。
在心肌炎、心包炎、冠心病、肺炎和炎症性肠病的患者中,AF 发生率较高,可能与炎性细胞浸润心肌引起氧化应激有关[18]。炎症激活肾素-血管紧张素-醛固酮系统,进而激活NADPH 和NOX。这些劣性刺激最终会激活TGF-β1信号转导,导致心肌结构重构和电重构。炎性因子和趋化因子可能与阵发性AF 向持续性AF 方向发展有关[18](图1)。
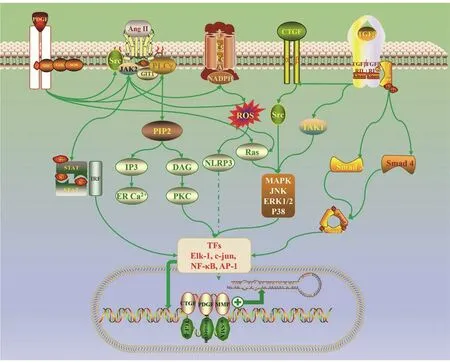
图1 纤维化所涉及的主要信号通路
3 miRNA
miRNA 是一种内源性、单链、短(17-22nt)、非编码核糖核苷酸。miRNA 在物种之间具有高度保守性,目前在植物、昆虫和哺乳动物中发现了数万种miRNA。根据miRBase(22.0 version)记载,已在人类鉴定出2 654 个成熟miRNA。
Dicer 酶是miRNA 合成过程中的关键酶之一,基因敲除小鼠和斑马鱼编码Dicer 的基因,发现具有胚胎致死性。而特异性敲除小鼠心肌中的Dicer基因会导致小鼠在幼年时期心力衰竭而死。在成年动物中,Dicer 酶活性降低会导致猝死和心肌肥厚[19]。因此,miRNA 对心血管病理生理过程起着重要作用,而miRNA 在AF 纤维化和电生理机制中已经有大量研究。
3.1 促纤维化miRNA
3.1.1miR-21 miR-21 作为纤维化研究最为广泛的miRNA 之一,在巨噬细胞中高度表达,它能促进巨噬细胞向M1 极化,而巨噬细胞通过旁分泌的方式作用于成纤维细胞,促进纤维化[20]。单细胞测序结果也显示antimiR-21 能减少心衰组织中成纤维细胞和巨噬细胞[21]。miR-21 的主要机制是通过抑制SPRY1 的表达,激活ERK/MAP 信号通路,导致成纤维细胞增殖[22]。miR-21 的过表达降低CADM1的表达,通过STAT3 信号通路促进心肌纤维化[23]。此 外,miR-21 的过表达也显著增强TGF-β1诱导的Smad7 下 调[24]。PTEN 已 被 证 明 是miR-21 的 靶 基因[25]。
3.1.2miR-23b-3p 和miR-27b-3p miR-23b-3p 和miR-27b-3p 位于人类9 号染色体上的miRNA 簇的单个转录单元内。通过靶向TGIF1 和PTEN 分别激活心脏成纤维细胞中的TGF-β1/Smad2/3 和AKT/N-钙黏蛋白信号通路,抑制miR-23b 可以缓解盲肠结扎诱导的晚期脓毒症模型中的心脏纤维化[26]。在心肌细胞中miR-27b 过表达可通过靶向PPAR-γ 促进心肌纤维化[27]。有研究表明TGFβR3抑制心脏成纤维细胞中TGF-β1表达、Smad2/3 激活以及胶原蛋白的沉积[28]。而TGFβR3 是miR-23b-3p 和miR-27b-3p 共同标靶[29]。
3.1.3其他促纤维化miRNA miR-10a 能通过TGF-β1/Smad 轴来调节成纤维细胞的增殖和分化[30]。在心肌特异性表达的miR-208 过表达会导致心肌纤维化,且miR-208a 表达上调仅限于心肌纤维化区域[31]。此外有研究发现miR-146b-5p 靶向作用于TIMP4 促进心肌纤维化[32]。
3.2 抗纤维化miRNA
3.2.1miR-29 miR-29 家族由两个双顺反子miRNA 簇合成的三个成员(miR-29a、miR-29b、miR-29c)组成。所有家族成员共享一个保守种子区,它们之间序列仅相差一个碱基。miR-29 是与纤维化相关性最强的miRNA 之一,并且优先在成纤维细胞中表达。弹性蛋白、原纤维蛋白1、Ⅰ型和Ⅲ型胶原都包含一个或多个miR-29 保守的潜在种子序列。在AngⅡ诱导的小鼠心肌纤维化模型中,miR-29b/c-3p 降低,而研究证实miR-29b/c-3p 靶向作用于TGF-β2和MMP-2[33]。此外,还有研究报道称miR-29 可 以 调 控DNMT3A[34]和VEGF/MAPK[35]来 抑制纤维化。但Sassi 等[36]得出相反结论,在病理性左室肥大的小鼠模型中,过表达miR-29 会促进心肌肥厚和纤维化,并且它能通过激活Wnt 信号通路促进心脏的病理性重塑。
3.2.2miR-30 miR-30 家族成员在大脑中高度表达,但miR-30c 除外,它在心脏成纤维细胞中含量丰富,并参与心肌纤维化[37]。在四氯化碳诱导的肝纤维化模型中,miR-30c 能调节肝星状细胞中TGF-β依赖性的细胞外基质相关基因的表达[38]。Wang等[39]提出miR-30c 可抑制肾纤维化并改善肾功能。在病理性左室肥大的模型中,miR-30c 能抑制心室胶原合成[37]。miR-30c 可能通过降低α-平滑肌肌动蛋白和波形蛋白的表达来抑制TGF-β1诱导的成纤维细胞向肌成纤维细胞分化,miR-30c 过表达会降低细胞增殖和迁移,miR-30c 可能通过靶向作用于TGFβR2 来减少心房纤维化[40]。
3.2.3miR-133 miR-133 在人类心肌中含量丰富。在大鼠心肌肥厚模型中,CTGF 的表达是对照组的5 倍,而靶标CTGF 的miR-133 和miR-30c 都 下调[41]。在成纤维细胞中过表达miR-133 可以通过抑制CTGF、降低胶原蛋白的产生[42]。基因敲除miR-133 小鼠会出现严重的纤维化和心力衰竭,并且增加猝死的概率[43]。荧光素酶报告基因检测证实TGF-β1和TGFβR2 是miR-133 和miR-590 的 靶标[44]。但最近的研究显示,缺氧/再灌注处理的人内皮祖细胞外泌体中miR-133 表达增加,并且促进心肌成纤维细胞的血管生成和内皮-间质转化[45]。
3.3 其他抗纤维化miRNAShan 等[44]通过尼古丁给药和建立快速起搏犬AF 模型中,发现尼古丁能够显著上调TGF-β1和TGFβR2 表达,降低miR-590 的水平。将miR-590 转染到犬心房成纤维细胞中会降低TGF-β1和TGFβR2 水平以及胶原蛋白含量,而antimiR-590 则消除这种保护作用。miR-132可能靶向作用于CTGF 表现出抗纤维化作用[46]。miR-499 下调会增加胶原蛋白沉积和心肌肥大[47]。
4 miRNA 与电生理重塑
AF 发生数小时内,就会发生离子通道的重构。其特征在于L 型钙离子通道电流(ICaL)和瞬时外向电流(Ito)显著下调,内向整流K+电流(IK)和乙酰胆碱依赖性K+电流(IKAch)上调。这些变化会缩短APD 和 有 效 不 应 期(effective refractory period,ERP),促进AF 的维持和发展。
4.1 miRNA 与钙稳态失衡L 型钙离子通道亚基α1(Cav1.2;CACNA1C)和L 型钙离子通道亚基β1和β2(Cavβ1/2;CACNB1/2)表达减少会导致ICaL降低和APD 缩短。当前研究显示miR-328 在AF 心房组织中变化存在争议。但体内转染miR-328 腺病毒的犬和过表达miR-328 的转基因小鼠中,Cav1.2,Cavβ1 和ICaL降低,APD 缩短并 增强AF 易感性,而antimiR-328 可逆转这一变化[48]。miR-21也能促进电重构,体外过表达miR-21 能降低ICaL密度,荧光素酶检测证实miR -21 靶向作用于CACNA1C 和CACNB2[49]。
较高的心房率会导致Ca2+超负荷和细胞内Ca2+稳态失衡,从而导致AF 向持续性方向发展。舒张期Ca2+通过雷诺定受体2(ryanodine receptor,RYR2)从肌浆网泄漏会增加Na+/Ca2+转运,使细胞膜去极化,缩短ERP。miR-106b-25 能抑制RYR2翻译,荧光素酶基因报告证实该簇的miR-93 直接靶向RyR2。此外,在miR-106b-25 基因敲除小鼠中,肌浆网Ca2+释放增加,AF 易感性明显增加[50]。胞质中的Ca2+通过2a 型肌浆/内质网钙离子三磷酸腺苷 酶(sarcoplasmic/endoplasmic reticulum Ca2+ATPase 2a,SERCA2a)转运,从而调节肌浆和胞质中Ca2+浓度。Cañón 等[51]发现AF 患者中miR-208b与SERCA2 表达呈负相关,而体外过表达miR-208b能降低SERCA2 蛋白的表达,增加AF 易感性。
4.2 miRNA 与K+通道IK对于AF 的发生发展起重要作用,编码内向整流K+通道的基因表达增加会增加IK,并且缩短APD,增加AF 易感性,但由于AF发生时所涉及的K+通道特别复杂,因此还需要进行进一步的深入研究。研究发现miR-30d 在AF 患者中高表达,而KCNJ3/Kir3.1 下调,通过荧光素酶报告基因发现miR-30d 直接靶向KCNJ3[52]。miR-1 是一种肌肉表达丰富的特异性miRNA。在兔心房快速起搏模型中,miR-1 靶向作用于KCNE1 和KCNB2,导 致ERP 缩 短,IK增 加,AF 易 感 性 增 加[53]。然而,Girmatsion 等[54]报道AF 患者心房组织中和离体人心房组织切片心动过速模型中miR-1 的水平显著降低,而IK上调。在犬和小鼠快速起搏模型中,心房组织和成纤维细胞中miR-26a/b 都降低[55]。在犬成纤维细胞中使用antimiR-26a 可导致IK增加、静息膜电位超极化并促进成纤维细胞增殖[55]。AF 患者心房中miR-26 与IK呈负相关,荧光素酶报告基因证实miR-26 直 接 靶 向KCNJ2[56]。此 外,miR-499 在AF 患者心房组织中上调,而KCNN3 表达下调,通过功能缺失和获得实验显示miR-499 与KCNN3 表达呈负相关。荧光素酶报告基因显示KCNN3 是miR-499 的标靶[57]。
5 miRNA 介导自主神经重塑
心肌电活动与自主神经和乙酰胆碱释放密切相关,它们功能失调对AF 的发生起着重要作用。在持续性AF 患者中,miR-30d 上调与IKAch下调有关[58]。miR-206 通过直接靶向超氧化物歧化酶的表达促进自主神经的重塑,从而增加ROS 的产生并缩短ERP。在miR-206 重组慢病毒转染的犬中,证实ROS 过量产生会增加AF 易感性[59]。在犬心房快速起搏模型中,miR-206 还可以通过靶向GCH1 促进自主神经重塑,从而增加AF 敏感性[60]。
6 miRNA 治疗局限性与展望
与传统疗法相比,miRNA 疗法优势之一是能够调节AF 纤维化和电生理重构过程中相关的多个靶基因或网络。在miRNA 表达降低的心血管疾病中,可以使用过表达策略来提高miRNA 水平。mimics-miRNA 是人工合成的编码成熟的miRNA序列,它是包含有引导链和互补过客链的双链寡核苷酸,能够形成RNA 诱导沉默复合体(RNA-induced silencing complex,RISC)。对于那些上调并导致疾病的miRNA,可以设计反义寡核苷酸,它与靶标miRNA 完全互补并降低其活性。由于天然核酸在生物环境中会被酶活性迅速降解,因此通过设计促进细胞渗透的胆固醇部分可以与反义寡核苷酸结合形成antago-miRNA,从而增强其稳定性。然而使用常规的lipofectamine、腺病毒或慢病毒将miRNA 转染至人体可能遭受未知的风险。因此,开发特殊的载体装载miRNA 显得尤为重要。
外泌体是直径为40~100 nm 的囊泡,几乎所有细胞都能分泌。外泌体纳米级的尺寸使得它们能够逃脱单核吞噬细胞的快速吞噬作用、通过血管内皮、穿越血脑屏障和胎盘屏障[61,62]。同时,由于它由特定的细胞分泌可作用于特定细胞,具有良好的靶向性。在生理状态下,miR-23/27b-3p/29/30/133都能包裹在外泌体中。但人工提取天然外泌体静脉注射可能面临全身过敏反应,以及外泌体聚集在肝脏组织中而失去效能等问题。辉瑞公司的新型冠状病毒mRNA 疫苗通过纳米脂质颗粒封装mRNA 似乎提供了部分解决方案。此外,类似外泌体的纳米脂质颗粒表面可以用识别靶细胞特定抗原的抗体包被,以此提高靶向性(图2)。
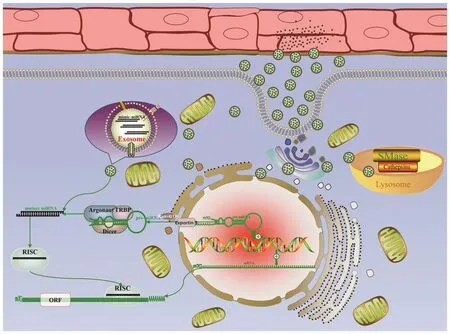
图2 装载mimics-miRNA 的外泌体和内源性miRNA发挥功能示意图
7 总结
大量数据表明,miRNA 对AF 的发病机理具有重要作用。随着高通量测序和单细胞测序的普及,miRNA 在AF 不同类型细胞中表达差异及作用机制将得到深入研究。各细胞间外泌体表面特异性配体的发现将会提高人工合成纳米脂质体的靶向性。它们为miRNA 抗心律失常治疗应用于临床提供了新的途径。
