RAD-seq技术研究鹅掌楸属种源遗传多样性和遗传结构*
2022-07-20潘文婷孙建军原勤勤张利利邓康桥厉月桥
潘文婷 孙建军 原勤勤 张利利 邓康桥 厉月桥
(中国林业科学研究院亚热带林业实验中心 分宜 336600)
木兰科(Magnoliaceace)鹅掌楸属(Liriodendron)为落叶大乔木,叶大形似马褂,故又称之为“马褂木”,欧洲人称其为“郁金香树”,是珍贵用材和优良观赏树种。现仅存两个种,即北美鹅掌楸(Liriodendrontulipifera)和鹅掌楸(Liriodendronchinense),为孑遗树种,前者生长于美国东部及加拿大南部的阔叶林中,从平原到山区呈连续分布,遗传资源十分丰富; 后者大多零星分布于我国长江流域以南的亚热带中、低山区,常混生于常绿或落叶阔叶林中(郝日明等, 1995),虽分布范围较大且遗传多样性良好,但因种群数量稀少,种群片段化严重,天然更新不良,处于濒危状态,已被列入我国二级珍稀濒危保护植物(王章荣, 2005)。鹅掌楸属中的这两个种是典型的东亚-北美间断分布“种对”(Vicariad Species Pairs)(Parksetal., 1990),是植物群体遗传学和分子系统发育地理学的理想材料(李康琴, 2013)。
随分子标记研究方法的增多,对鹅掌楸分子育种研究也逐渐完善,王晓阳等(2011)利用SSR技术研究鹅掌楸苗期生长杂种优势; 李康琴(2013)基于SSR技术对鹅掌楸属进行群体遗传结构及分子系统地理学研究; 罗群凤等(2015)采用同源克隆和RACE技术克隆北美鹅掌楸的查尔酮合成酶基因(LtCHS),并对其进行生物信息学及组织表达进行了分析; 祁荔(2017)利用AFLP及SSR分子标记构建鹅掌楸遗传图谱及重要性状QTL初步定位。近年来基于全基因组酶切位点相关的简化基因组测序基础上发展出来了二代测序技术RAD-seq (Restriction-site associated DNA sequencing),该技术由Miller等(2007)提出,主要利用酶切、序列捕获降低基因组复杂度从而获得部分基因组序列信息(Wangetal., 2017; Jérémyetal., 2020),作为一种快速有效的手段,实现大规模的分子标记开发,既节约成本又节省时间,已成功应用于 SNP 标记的开发、动植物重要经济性状的 QTL 定位、生物进化等研究领域(Fengetal., 2020; Lietal., 2020)。陆叶等(2019)利用RRAD-seq 技术开展了鹅掌楸基因组 SNP 标记开发,最终获得3 501个候选SNP标记; Sheng 等(2021)利用RAD-seq技术分析了鹅掌楸Myb基因家族的表征及其在非生物胁迫应答中的作用,可见RAD-seq技术能提供更多SNP标记,有效补充了鹅掌楸遗传信息较少这一问题,但有关鹅掌楸属群体遗传结构和遗传分化的研究较少。
开展群体结构和遗传分化的研究能精准评估遗传多样性和制定科学有效的种质资源保护策略,李云飞等(2019)采用RAD-seq技术开发了85种杜鹃花属植物的高质量SNP位点,通过高通量测序探讨分类,在亚属水平的分类取得很好的效果。黄承玲等(2021)采用 RAD-seq技术对34种杜鹃花属(Rhododendron)植物进行分类,证实了RAD-seq技术在复杂植物类群的物种分类方面比传统分子标记具有明显优势。本研究利用RAD-seq测序技术,以9个中国鹅掌楸和4个北美鹅掌楸种源为研究材料,通过高通量测序,对13个鹅掌楸属种源143份样本进行了遗传多样性、遗传变异、遗传分化以及群体基因交流等分析,揭示鹅掌楸属各种源亲缘关系,以期为鹅掌楸属分子鉴定、种质创新及种质资源收集与保存提供参考。
1 材料与方法
1.1 试验材料
试验材料共有13个鹅掌楸属种源,其中9个来自中国7个省,另外4个来自美国,相关种源地理信息详见表1。所有种源种植在中国林业科学研究院亚热带林业实验中心年珠实验林场鹅掌楸种源试验林中。分别于2016年10月和2017年5月进行采样,每个种源随机采取10~14株树,每株树取5~6片叶,共采集了143份鹅掌楸属样本,其中有97个样本采自9个中国鹅掌楸种源,还有46个样本采自4个北美鹅掌楸种源。样本用自封袋装好并倒入硅胶干燥、保存。
1.2 基因组DNA的制备
全部143份鹅掌楸属样本的基因组DNA采用改良的CTAB法(Liuetal., 2008)提取,用 1.0%琼脂糖凝胶检测DNA质量。
1.3 酶切建库
利用RAD-seq技术对143份鹅掌楸属样本的基因组DNA进行简化测序。采用限制性内切酶AvaII与MspI酶切组合对全基因组DNA完全酶切(Petersonetal., 2012),(插入片段范围: 500~600 bp,将酶切产物进行5’末端修复,同时对5’末端进行磷酸化修饰,3’末端加A,使之与接头5’端T互补,提高接头连接效率,阻止接头自连。然后连接测序接头,将连接产物锚定在flowcell上,进行桥式扩增。用琼脂糖凝胶电泳进行片段大小选择,通过PCR扩增,增大文库量,建好的文库用Illumina HiSeq X10进行测序。

表1 各参试种源地理信息及参试数量Tab.1 Geographic information and sample number of 13 provenances in the experiment
1.4 统计分析
对原始数据进行识别、过滤、质量评估,再进行单样本测序数据的 RAD 标记开发、样本群体的RAD标记开发、多态性标记SNPs开发等,最后开展群体多样性分析,包括进化树构建及群体结构分析等。
主要通过Stacks软件(http:∥creskolab.uoregon.edu/stacks/)(Catchenetal., 2013)进行RAD标记开发。基因组经酶切处理打断为多个小片段,每个片段相当于一个标记位点,同一位点的序列通过相似性聚类,形成一个Stack。每个Stack中存在数条高深度片段,其余全为低深度片段。高深度片段即为潜在基因型,低深度片段可能由于测序错误导致。
采用Mega5.1软件(http:∥www.atgc-montpellier.fr/phyml/)(Tamuraetal., 2011)中的Upgma算法和Neighbour-joining分别构建13个种源的关系树和143份样本的进化树。通过 Admixture软件(Lexanderetal., 2009)对143份样本的群体结构进行聚类分析,该软件运算速度快,创建Plink(http:∥pngu.mgh.harvard.edu/~purcell/plink/)的输入文件,用Plink进行SNP过滤,将样品的分群数(K值)设定为1-6进行聚类,根据CV error最小值确定分群数。
使用POPGENE 1.32软件(http:∥www.ualberta.ca/~fyeh/download.htm.)(Yehetal., 2000)计算观察杂合度(Ho)、期望杂合度(He)、核苷酸多样性(π)和基因分化系数(Gst)等主要遗传结构的度量参数。
2 结果与分析
2.1 RAD-seq测序及质量评估
本研究通过RAD-seq测序技术,完成13个鹅掌楸属种源的143份样本的测序,总碱基数为396.95 Gb,样本平均获取的碱基数为2.76 Gb; 测得有效数据为1 323.17 Mb,样本平均获取的有效数据为9.21 Mb; 共开发RAD标签 2 611 618个,样本的平均标签数为141 222个,平均测序深度为25.02 x(表2)。所测序列GC含量较低(平均值为26.51%),Q30数据较高(平均值为95.07%),表明错误率低,测序结果可靠。通过对样本间进行多态性分析,共得到高质量的SNP位点4 454个。

表2 RAD-seq测序数据统计Tab.2 Statistics of RAD-seq sequencing data of 13 provenances
2.2 种源遗传多样性统计
对13个鹅掌楸属种源的遗传多样性相关参数进行统计(表3),结果表明,Ho为0.013 2~0.049 6,均值为0.037 9;He为0.022 2~0.059 7,均值为0.047 7;π为0.023 6~0.064 0,均值为0.050 6。采用隶属函数法(彭松等, 2014)对Ho、He和π进行综合评价(表4),其中遗传多样性排前三位的种源依次为EX,NK和BK,DBS种源的遗传多样性最低。
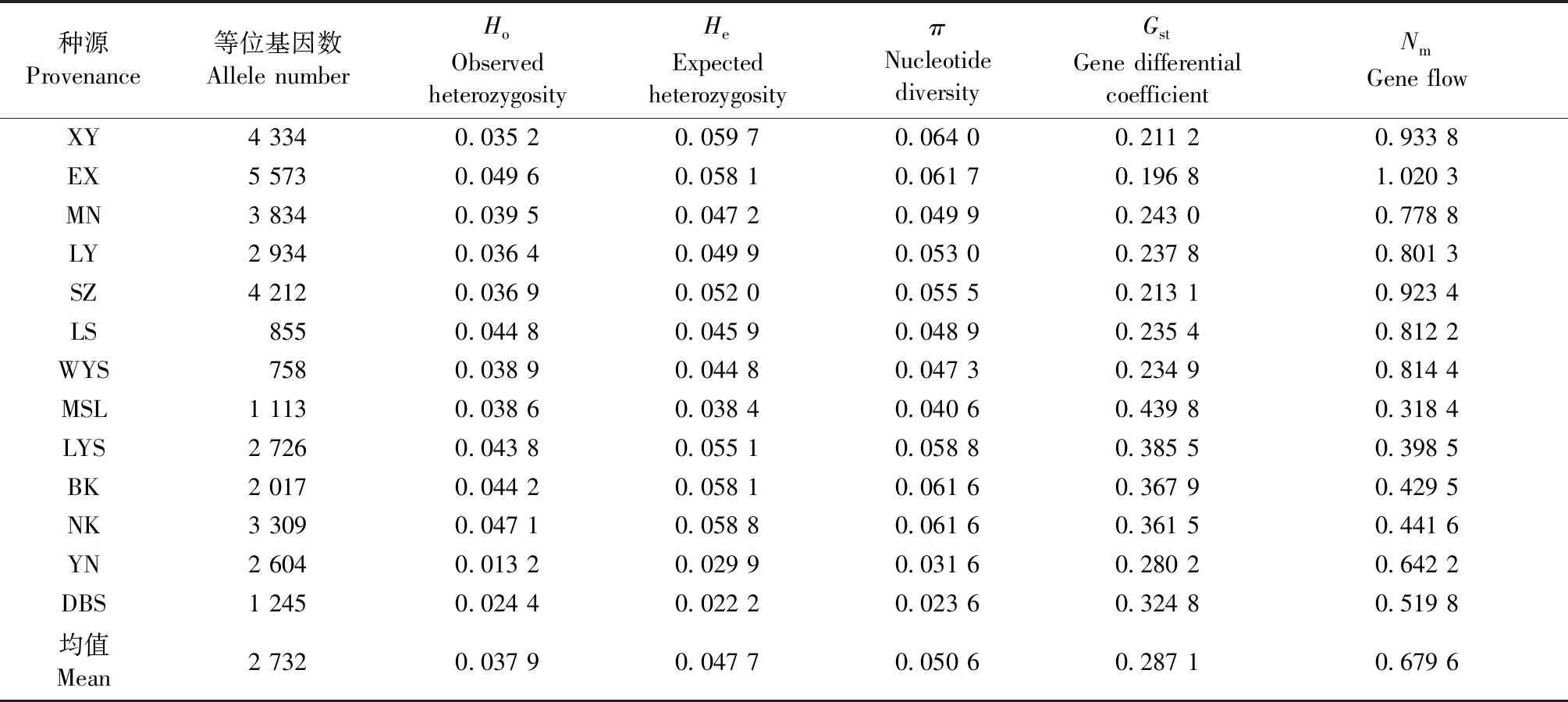
表3 各种源遗传结构参数统计Tab.3 Statistics of genetic structure in different provenances

表4 13个鹅掌楸属种源遗传多样性各指标的隶属函数值Tab.4 Subordinative function values of genetic diversity parameters among 13 provenances in Liriodendron
2.3 遗传结构分析
对13个鹅掌楸属种源的Gst和Nm数据(表3)进行分析,结果表明Gst从0.196 8到0.439 8,鹅掌楸种源之间存在较大的遗传分化(Gst=0.241 9)以及中等的基因流(Nm=0.805 1,其中种源EX的Nm>1),北美鹅掌楸种源之间存在很大的的遗传分化(Gst=0.388 6>0.25)以及低水平的基因流(Nm=0.397 0),表明北美鹅掌楸遗传变异主要存在于种源间。
基于筛选的有效SNP,通过Mega5.1 软件中的Upgma 算法和Neighbour-joining分别构建13个种源的关系树(图1)和143份样本的进化树(图2),结果显示,13个鹅掌楸属种源被分为3个类群,其中XY、YN、EX、SZ和MN 5个种源为一个类群; LY、DBS、LS和WYS 4个种源为一个类群; NK、LYS、MSL和BK 4个种源为一个类群。
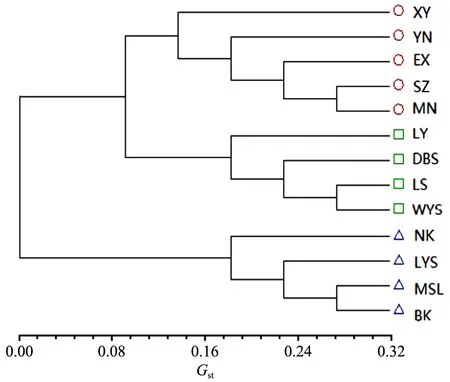
图1 13个种源的关系树Fig. 1 Relation tree of 13 provenances in Liriodendron
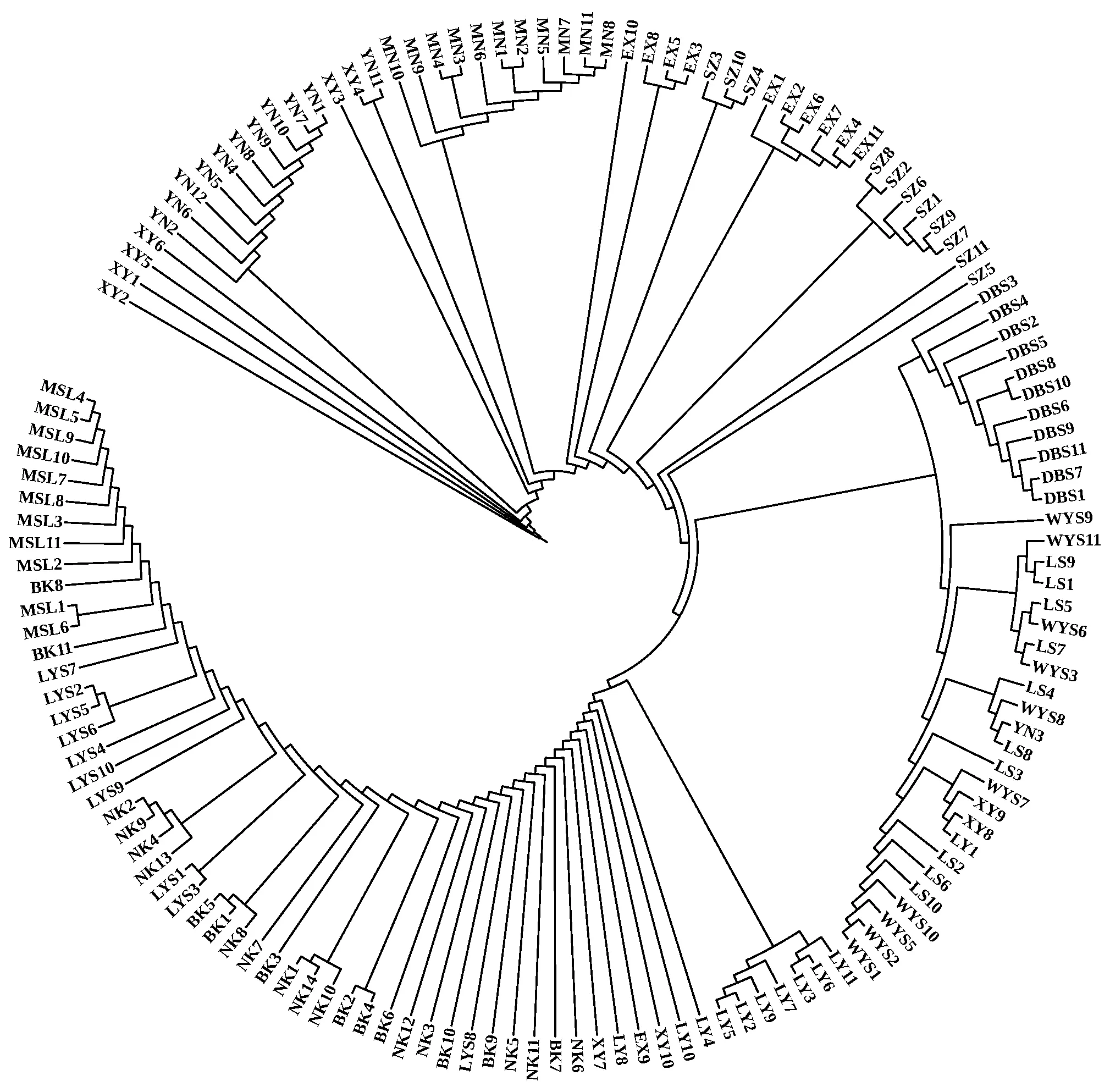
图2 143份样本的进化树Fig. 2 Neighbour-joining phylogram illustrating genetic relationships among 143 individuals每一个分枝代表一份种质材料,树枝长度代表两个物种间的进化距离,树分支上的数字代表该分支的支持的百分数。Each branch represents one piece of germplasm, and the length of the branch represents the evolutionary distance between two species, the number on a branch of the tree represents the percentage of support for that branch.
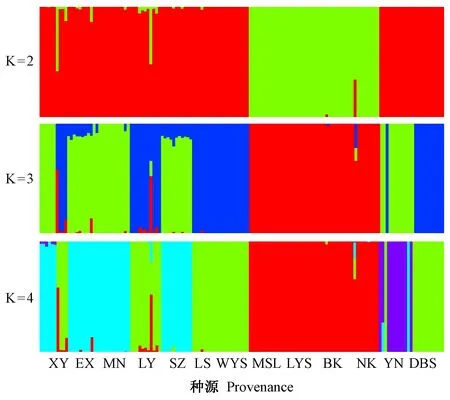
图3 分群数为2~4的聚类图Fig. 3 Bar plot of 2-4 clusters identified with adegenet R package左图图中每种颜色代表一个群体,每列代表一个样品的情况; 图中展示了143个样品分群值从2~4的聚类情况。右图中为每个K值对应的CV error值,K为3的时候最小。The picture left shows the clustering of 143 samples with cluster values from 2 to 4 (each color represents a subgroup,Each column represents a sample). The figure on the right shows the CV error value corresponding to each K value (When K is 3, the CV error value is minimal).
通过Admixture 软件对143份样本的群体结构进行聚类分析,假设样品的分群数(K值)为1~6,对应的CV error值分别为: 0.465 4,0.178 6,0.142 1,0.153 1,0.163 2,0.168 9(图3)。其群体结构最优分群数为3,分群的结果与关系树和进化树分析所得结果基本一致。
3 讨论
本研究通过RAD-seq测序技术,完成13个鹅掌楸属种源的143份样本的测序,平均测序深度为25.02 ×,共获得总碱基数396.95 Gb,平均每个样本的碱基数为2.76 Gb,获得高质量SNPs标记4 454个。比李云飞等(2019年)获得的3 501个SNP 标记多,这与本研究有13个种源的研究材料,而李云飞等(2019年)为2个种源有关,但均比以往鹅掌楸的分子标记开发技术获得的候选SNPs数量有显著提高。这些标记进一步增加了鹅掌楸属的基因组资源量,可更准确地界定群体分类,为鹅掌楸属资源的遗传保存、育种及改良应用提供依据(李云飞等, 2019; 黄承玲等, 2021)。
以获得的高质量SNP位点为基础,进一步分析13个鹅掌楸属遗传多样性和遗传结构。遗传多样性排前三位的种源依次为EX,NK和BK,DBS 种源的遗传多样性最低。但总体与报道的鹅掌楸遗传多样性数值(李康琴, 2013; 赵亚琦等, 2014)相比较低,可能是由于遗传多样性的估算受样本数量、采样策略及基因分型数据等因素的影响,本研究样本数量较少,且为种源试验林内采样,因此遗传多样性较低。
对13个鹅掌楸属种源的Gst和Nm数据进行分析,结果表明Gst从0.196 8到0.439 8,鹅掌楸种源之间存在较大的遗传分化以及中等的基因流(Gst=0.241 9、Nm=0.805 1,其中种源EX的Nm>1),北美鹅掌楸种源之间存在很大的的遗传分化以及低水平的基因流(Gst=0.388 6>0.25、Nm=0.397 0),表明鹅掌楸属遗传分化变异主要存在于种源间; EX种源的Nm值大于1,表明基因流可以防止由遗传漂变引起的群体间的遗传分化,但各种源间Nm均值只有0.679 6; 且多数种源Ho小于期望杂合度He。群体间的高度分化的根源在于由生境片段化而引起的遗传漂变和群体间低水平的基因流(Zaoualietal., 2012),可见鹅掌楸属存在由小群体效应和片段化影响导致的濒危现象,该结论与李康琴(2013)的研究的结果一致。
通过对13个鹅掌楸属种源的143份样本进行聚类分析和群体结构分析,可将13个鹅掌楸属种源分为3个类群,其中 XY、YN、EX、SZ和MN 5个种源为类群1; LY、DBS、LS和WYS 4个种源为类群2; NK、LYS、MSL和BK 4个种源为类群3。经过分析可见类群1和类群2分别处在中国西部和东部,该结论进一步验证了朱晓琴等(1995)和李康琴(2013)的研究结果,可见鹅掌楸属遗传结构的形成与其地理隔离和片段化分布有关。中国东部种源群(类群2)较中国西部种源群(类群1)遗传多样性高(Ho: 0.036 1>0.034 9),但均低于北美鹅掌楸(类群3)遗传多样性,该结论与李康琴(2013)的结论一致。进化树和聚类图均显示3个类群间出现了少数的个体混杂现象,表明群体间有基因交流的现象,因此,利用这些材料进行杂交育种时,既要考虑其亲缘关系也要注意他们的遗传结构。
4 结论
采用RAD-seq技术对鹅掌楸属13个种源143份样本进行测序,获得了高质量SNP位点,进而分析鹅掌楸属遗传多样性、群体结构分析及进化树的构建,鹅掌楸属13个种源被分为3个类群,其中北美鹅掌楸和鹅掌楸可显著区分、中国东部鹅掌楸和西部鹅掌楸的差异也很显著。本研究结果表明高效的RAD-seq技术获得的大量SNP位点可应用于鹅掌楸属物种的区分,进一步证实了鹅掌楸属遗传结构的形成与其地理隔离和片段化分布有关,且存在由于小群体效应和片段化影响导致的濒危现象; 同时这些SNP标记可进一步用于鹅掌楸属QTL定位、遗传连锁图谱、分子辅助育种等研究,进而为鹅掌楸属的遗传演化研究提供理论基础,为今后鹅掌楸属种质创新及种质资源收集与保存提供参考依据。
