利用三中心氢键限制芳酰胺构象的核磁共振氢谱分析
2022-07-18闵婧王力彦
闵婧, 王力彦
利用三中心氢键限制芳酰胺构象的核磁共振氢谱分析
闵婧, 王力彦
(吉林大学化学学院, 超分子结构与材料国家重点实验室, 长春 130012)
设计并合成了9个可形成三中心氢键和6个可形成二中心氢键的-芳基芳酰胺模型化合物, 基于它们在氯仿和DMSO中的一维核磁共振波谱, 系统地分析了羰基对H和H的去屏蔽效应. 将Δ(δH)和Δ(δH)的值结合在一起, 分析了三中心氢键对芳酰胺分子的构象限制效果, 发现-(2-氟苯基)-2-氟苯甲酰胺、-(2-甲氧基苯基)-2-氟苯甲酰胺和-(2-氟苯基)-2-甲氧基苯甲酰胺这3个-芳基芳酰胺在酰胺基团的左右两侧都能展现出很好的构象控制效果, 因此认为这3种结构单元在构建折叠体方面具有更大的潜力. 此外, 本文还发现, 当NH与第二个氢键受体形成氢键时, 其和第一个氢键受体之间的氢键就被削弱了, 即在芳酰胺形成三中心氢键时, 2个氢键受体争相与NH形成氢键并取得了某种平衡.
三中心氢键; 芳酰胺; 构象; 折叠体
分子内氢键在有机分子和生物大分子形成稳定构象、 产生特定三维结构的过程中常起到至关重要的作用[1], 形成分子内氢键可能改变分子的水溶性、 亲脂性和膜渗透性等物理化学性质[2]. 由一个氢键给体和一个氢键受体组成的二中心氢键[1]是最常见的氢键结构[3~5]. 而三中心氢键是由一个氢键给体与两个氢键受体同时结合形成的[6~9], 如果氢键给体是XH, 那么H原子将接近X与两个受体原子形成的平面, 因此三中心分子内氢键可以用于限制分子内旋转[1].
近30年来, 化学家们发展了许多具有不同构象和分子结构的芳酰胺折叠体[3,10~12], 其中往往包含能够形成三中心分子内氢键的结构基元[13~16]. 1994年, Hamilton等[17]首先报道了一系列具有折叠构象的邻氨基苯甲酸低聚物(Oligoanthranilamides), 其中三中心氢键对于构象的形成起到了非常重要的作用. 此后, 一些研究组发展了由苯、 吡啶和喹啉等结构基元构筑的许多折叠体[18~23]. 2005年, Li等[19]利用N—H…F氢键诱导芳酰胺形成螺旋结构, 并在晶体结构中观察到三中心分子内氢键. Jiang等[24,25]报道了含有8-氨基喹啉结构基元的低聚酰胺折叠体, 晶体学研究表明四聚体通过三中心氢键形成了单螺旋. 红外光谱[14]、 X射线衍射[22,26]和核磁共振波谱都是常用的表征氢键的方法. 而在溶液中表征氢键主要依赖于二维核磁共振波谱、 变温核磁共振波谱及氢氘交换等实验. 例如龚兵等利用二维核磁共振波谱技术检测到酰胺质子与相邻的烷氧基质子之间强烈的NOE效应, 证明了三中心氢键和折叠构象的形成[18]. Hamilton等[27]通过一维核磁共振波谱的变温实验证明酰胺质子形成了三中心氢键. 黎占亭和张文科等[28]采用氢氘交换实验测定了酰胺发生交换的速率, 评估了折叠体内不同酰胺NH形成三中心氢键的能力. 此外, 近年来原子力显微镜[29,30]也被应用于氢键的直接观察. 由于羰基具有磁各向异性, 接近羰基平面的C—H质子表现出显著的去屏蔽(+)效应[31,32]. 基于此, 本课题组分析了一系列-烷基芳酰胺以及3个-芳基芳酰胺的核磁共振氢谱, 建立了一种使用位于芳酰胺羰基位的质子的化学位移变化来判断芳酰胺构象, 进而分析分子内氢键的方法, 称之为Δ(δH)方法[32].
本文设计并合成了9个可形成三中心氢键和6个可形成二中心氢键的-芳基芳酰胺模型化合物(Scheme 1), 基于它们在4种溶剂中的一维核磁共振氢谱, 系统分析了羰基对H和H的去屏蔽效应, 并结合Δ(δH)与Δ(δH), 讨论了各种芳酰胺亚结构单元形成平面构象的可能性. 这些结果将有助于芳酰胺折叠体的结构设计和开发.

Scheme 1Structures of⁃aryl aromatic amides
CO⁃O: 2⁃Methoxy⁃⁃(2⁃methoxyphenyl)benzamide; CF⁃O: 2⁃fluoro⁃⁃(2⁃methoxyphenyl)benzamide; CPy⁃O:⁃(2⁃methoxyphenyl)⁃2⁃pyridinecarboxamide; CPh⁃O:⁃(2⁃methoxyphenyl)benzamide; CO⁃F:⁃(2⁃fluorophenyl)⁃2⁃methoxybenzamide; CF⁃F: 2⁃fluoro⁃⁃(2⁃fluorophenyl)benzamide; CPy⁃F:⁃(2⁃fluorophenyl)⁃2⁃pyridinecarboxamide; CPh⁃F:⁃(2⁃fluorophenyl)benzamide; CO⁃Es: methyl 2⁃[(2⁃methoxybenzoyl)amino]⁃benzoate; CF⁃Es: methyl 2⁃[(2⁃fluorobenzoyl)amino]benzoate; CPy⁃Es: methyl 2⁃[(2⁃pyridinylcarbonyl)amino]benzoate; CPh⁃Es: methyl 2⁃benzoylaminobenzoate; CO⁃Ph: 2⁃methoxy⁃⁃phenylbenzamide; CF⁃Ph: 2⁃fluoro⁃N⁃phenylbenzamide; CPy⁃Ph:⁃phenyl⁃2⁃pyridinecarboxamide.
1 实验部分
1.1 试剂与仪器
2-甲氧基苯甲酸、 2-氟苯甲酸、 2-吡啶甲酸、 苯甲酸、 2-甲氧基苯胺、 2-氟苯胺、 氨茴酸甲酯、 苯甲酰苯胺、,′-羰基二咪唑和氯化亚砜等购于安耐吉试剂公司, 使用前未经进一步处理; 氘代氯仿(CDCl3)、 氘代乙腈(CD3CN)和氘代二甲基亚砜(DMSO-d6)购于CIL试剂公司; 氘代硝基甲烷(CD3NO2)购于Acros试剂公司; 苯胺、 碳酸氢钠、 无水硫酸镁、 三乙胺和氯化钠购于北京国药集团化学试剂有限公司; 二氯甲烷、 氯仿、 正己烷、 乙酸乙酯、,-二甲基甲酰胺(DMF)和四氢呋喃(THF)等购于天津富宇化工公司; 柱层析所用硅胶为FCP 200~300, 购于青岛鼎康硅胶有限公司.
Bruker AVANCEIII 500 MHz型核磁共振波谱仪(NMR), 德国Bruker公司.
1.2 实验过程
在核磁测试前, 将待测化合物置于50 ℃的真空烘箱中干燥12 h. 以CDCl3, CD3CN, CD3NO2和DMSO-d6为溶剂配制核磁测试溶液. 为了避免分子聚集对化学位移的影响, 测试溶液的浓度控制在(1.05±0.10) mmol/L范围内. 设定采样温度为298 K. 质子的化学位移参考核磁测试时溶剂残留峰的化学位移值(CDCl3,7.26; CD3NO2,4.33; CD3CN,1.94; DMSO-d6,2.50), 四甲基硅烷(TMS)在CDCl3和DMSO-d6中的信号介于-0.01~+0.01之间.
2 结果与讨论
为了系统地分析芳酰胺的构象, 合成了15个-芳基芳酰胺化合物(Scheme 1, 合成方法和表征见本文支持信息). 其中, CO-O, CF-O, CPy-O, CO-F, CF-F, CPy-F, CO-Es, CF-Es和CPy-Es是9个可形成三中心氢键的模型化合物, CPh-O, CPh-F, CPh-Es, CO-Ph, CF-Ph和CPy-Ph是6个可形成二中心氢键的模型化合物. 为了便于区别每个化合物芳香酸部分的质子和苯胺部分的质子, 将苯胺部分的质子用Hb表示, 例如CF-O中的H6b. 本文所关注的H和H是按照相对于羰基的位置, 它们在空间上与羰基接近. 由Scheme 2可见, 以CF-O为例,H位于2-氟苯甲酸的6位碳原子上(称为H6),H位于2-甲氧基苯胺的6位碳原子上(称为H6b), 因为NH形成了三中心氢键, 所以这两个质子处于羰基的去屏蔽区. 利用我们建立的基于羰基磁各相异性效应的构象分析方法[33], 可以独立地分析芳酰胺左右两侧的构象: 用Δ(δH)分析左侧的构象, 用Δ(δH)分析右侧的构象.
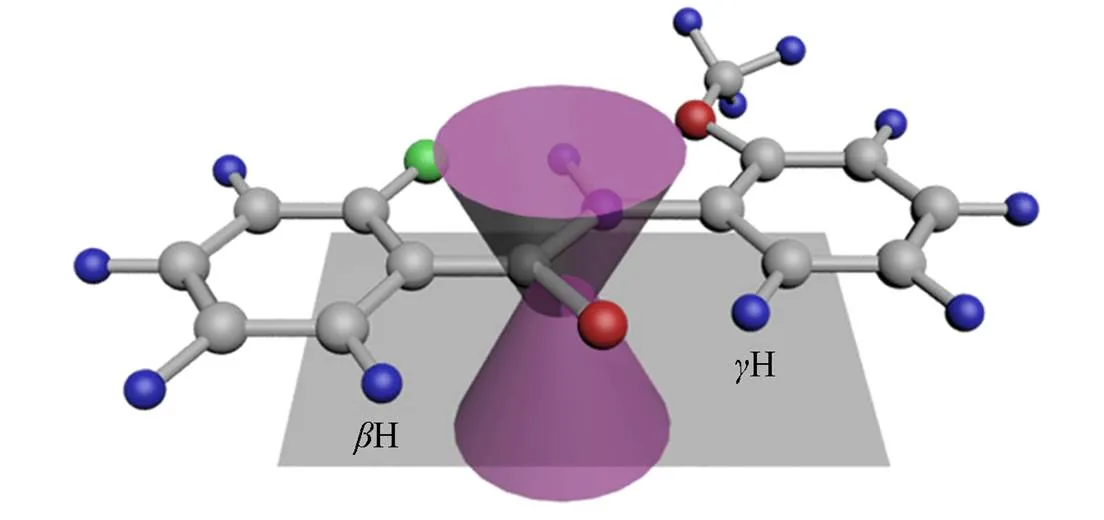
Scheme 2Schematic drawing of the magnetic anisotropy of the carbonyl group of the aromatic amide that can form a three⁃center hydrogen bond
Magenta cones represent the shielding region of the carbonyl group. Grey square represents the plane of the C=O double bond, in which protons,H andH, display deshielding effects in NMR spectra.
将这些-芳基芳酰胺化合物分别溶解在CDCl3、 CD3NO2、 CD3CN和DMSO-d6中, 记录并分析它们的1H NMR谱(图S1~图S13, 见本文支持信息, CO-O和CF-F的相关数据已经被报道[33]). 另外, 在本文中没有包括含有氨基吡啶基团的芳酰胺化合物的相关研究, 因为在角度和键长方面吡啶与酰胺NH不适合形成分子内氢键[2].
2.1 含有2-甲氧基苯胺基团的三中心氢键模型化合物
首先对比了3个含有2-甲氧基苯胺基团的三中心氢键模型化合物的H在不同溶剂中的化学位移及与溶剂相关的化学位移变化Δ(δH). 由表1可见, CF-O在氯仿和DMSO中化学位移的差值Δ(δH)-1为0.31, 表明羰基对H的去屏蔽效应比较明显[参考表S1(见本文支持信息)中除H,H外的芳香质子的Δ(ArH)-1的数值], 因此我们认为CF-O的酰胺NH与苯甲酰部分的氟在氯仿中形成了分子内氢键, N—H…F氢键对苯甲酰部分的构象限制能力较强. 而CO-O的Δ(δH)-1值为0.20, 表明酰胺NH与苯甲酰中甲氧基在氯仿中形成的N—H…O氢键对苯甲酰部分的构象限制能力不强. CPy-O的Δ(δH)-1则相对较小(0.10), 羰基对吡啶环上质子的去屏蔽效应不显著. 在考察H的化学位移时, 可以把CPh-O看作是CO-O, CF-O和CPy-O的参比分子, 它的Δ(δH)-1是绝对值很小的负值.

Table 1 Chemical shifts of βH of CO-O, CF-O, CPy-O and CPh-O in CDCl3, CD3NO2, CD3CN and DMSO-d6 and their solvent-related changes*
* Δ(δH)-1=δH(CDCl3)-δH(DMSO), Δ(δH)-2=δH(CD3NO2)-δH(DMSO), Δ(δH)-3=δH(CD3CN)-δH(DMSO).
我们总结了CO-O, CF-O和CPy-O的H在4种溶剂中的化学位移及它们溶剂相关的Δ(δH). 由表2可见, CF-O的Δ(δH)-1值最大(0.43), 去屏蔽效应比较明显.由表1和表2可见, 对于这组化合物, 只有CF-O的δH和δH同时表现出明显的去屏蔽效应, 表明CF-O基于三中心氢键形成了较好的平面分子构象. CPh-O的3个Δ(δH)的数值都很大, 说明二中心氢键对苯胺一侧的构象限制很有效, 同时暗示了在苯胺一侧构象限制的效果上, NH与两个氢键受体形成的三中心氢键不如NH只与苯胺环上的烷氧基形成的二中心氢键.
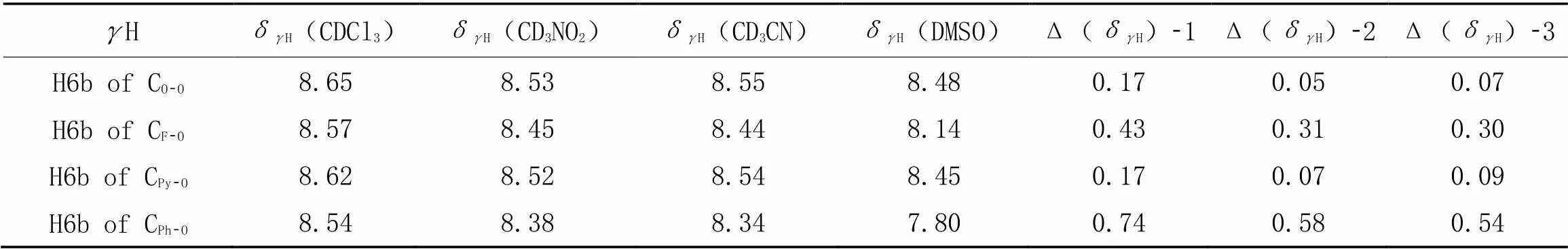
Table 2 Chemical shifts of γH of CO-O, CF-O, CPy-O and CPh-O in CDCl3, CD3NO2, CD3CN and DMSO-d6 and their solvent-related changes*
*Δ(δH)-1=δH(CDCl3)-δH(DMSO), Δ(δH)-2=δH(CD3NO2)-δH(DMSO), Δ(δH)-3=δH(CD3CN)-δH(DMSO).
2.2 含有2-氟苯胺基团的三中心氢键模型化合物
分析了Scheme 1中3个含有2-氟苯胺基团的三中心氢键模型化合物的H在4种溶剂中的化学位移及与溶剂相关的化学位移变化Δ(δH). 如表3所示, CO-F和CF-F的Δ(δH)-1值分别是0.35和0.47, 表明它们的酰胺NH与苯甲酰部分的烷氧基/氟形成了分子内氢键, 而且CO-F的N—H…O氢键和CF-F的 N—H…F氢键对构象的限制效果都很好.
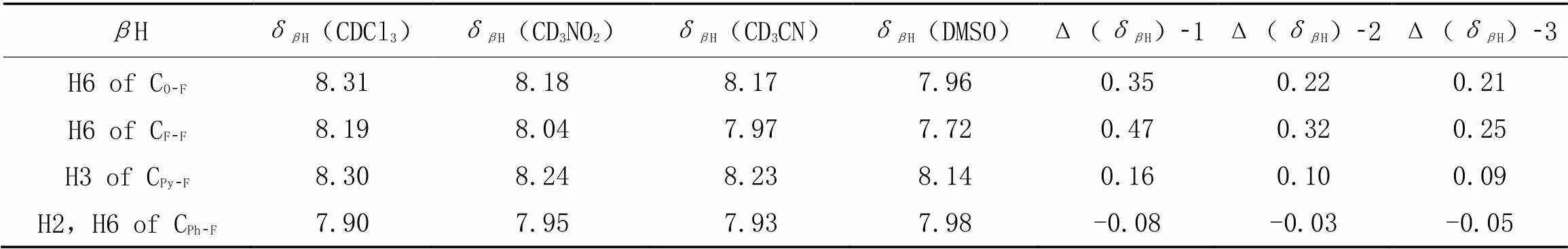
Table 3 Chemical shifts ofβH of CO-F, CF-F, CPy-F and CPh-F in CDCl3, CD3NO2, CD3CN and DMSO⁃d6 and their solvent-related changes*
*Δ(δH)-1=δH(CDCl3)-δH(DMSO), Δ(δH)-2=δH(CD3NO2)-δH(DMSO), Δ(δH)-3=δH(CD3CN)-δH(DMSO).
这组芳酰胺化合物的δH都表现出显著的去屏蔽效应. 由表4可见, CF-F的Δ(δH)-1值为0.72, 其表现出强烈的去屏蔽效应; CO-F和CPy-F的Δ(δH)-1值分别为0.38和0.40, 去屏蔽效应也很显著. 结合表3和表4结果可以看出, CF-F和CO-F的δH和δH同时表现出明显的去屏蔽效应, 表明这两个分子可以在多种溶剂中基于三中心氢键形成较好的平面分子构象.

Table 4 Chemical shifts of γH of CO-F, CF-F, CPy-F and CPh-F in CDCl3, CD3NO2, CD3CN and DMSO-d6 and their solvent-related changes*
*Δ(δH)-1=δH(CDCl3)-δH(DMSO), Δ(δH)-2=δH(CD3NO2)-δH(DMSO), Δ(δH)-3=δH(CD3CN)-δH(DMSO).
2.3 含有氨茴酸甲酯基团的三中心氢键模型化合物
表5总结了Scheme 1中3种含有氨茴酸甲酯基团的三中心氢键模型化合物的H在4种不同溶剂中的化学位移及溶剂相关的化学位移变化Δ(δH). CO-Es和CF-Es的Δ(δH)-1分别是0.19 和0.16 , 据此我们认为酰胺NH与它们苯甲酰部分的甲氧基和氟形成的分子内氢键具有限制构象的能力. 另外, CPy-Es的Δ(δH)-1值相对较小(0.10), 而且CPy-O和CPy-F的Δ(δH)-1值也都较小, 可能意味着Δ(δH)方法不适合分析含有吡啶甲酸基团的芳酰胺的分子内氢键[33].
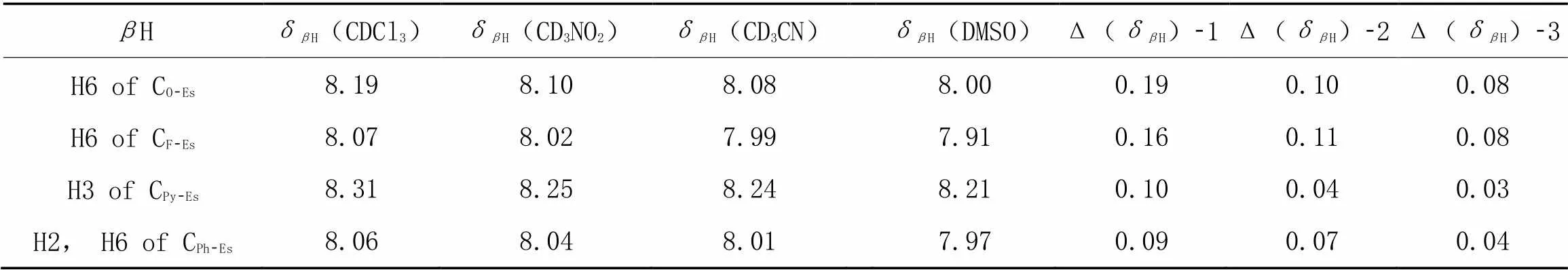
Table 5 Chemical shifts of βH of CO-Es, CF-Es, CPy-Es and CPh-Es in CDCl3, CD3NO2, CD3CN and DMSO-d6 and their solvent-related changes*
*Δ(δH)-1=δH(CDCl3)-δH(DMSO), Δ(δH)-2=δH(CD3NO2)-δH(DMSO), Δ(δH)-3=δH(CD3CN)-δH(DMSO).
对于含有氨茴酸甲酯基团的芳酰胺化合物, 羧酸甲酯与酰胺N—H经常形成的是N—H…O=C分子内氢键, 而N—H…O—CH3分子内氢键比较少见[2]. 表6总结了CO-Es, CF-Es和CPy-Es的H在4种不同溶剂中的化学位移及溶剂相关的化学位移变化Δ(δH). CF-Es的Δ(δH)-1值为0.34, 明显大于CO-Es和CPy-Es的Δ(δH)-1值(分别为0.17和0.12). 表明CF-Es的苯胺部分在氯仿中形成了较好的平面构象.
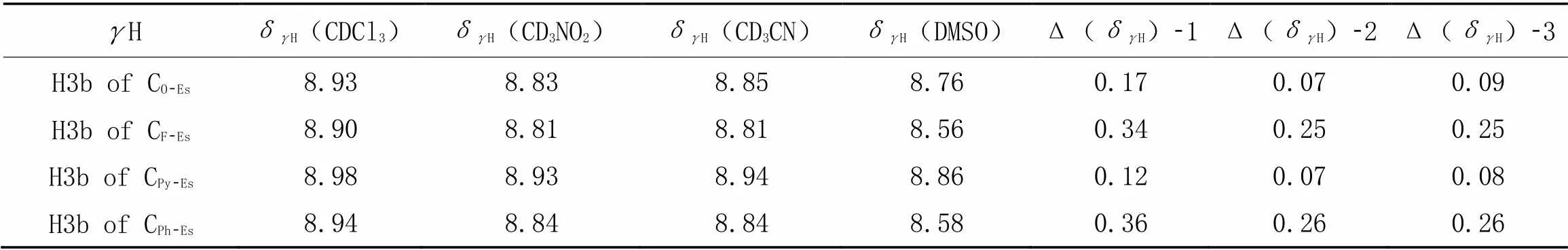
Table 6 Chemical shifts of γH of CO-Es, CF-Es, CPy-Es and CPh-Es in CDCl3, CD3NO2, CD3CN and DMSO-d6 and their solvent-related changes*
*Δ(δH)-1=δH(CDCl3)-δH(DMSO), Δ(δH)-2=δH(CD3NO2)-δH(DMSO), Δ(δH)-3=δH(CD3CN)-δH(DMSO).
2.4 二苯甲酰肼和二苯基脲的衍生物
除了-芳基芳酰胺模型化合物, 我们还合成了3个二苯甲酰肼衍生物, HO-O, HF-F和HF-O(Scheme 3), 它们的酰肼NH能够同时与羰基和甲氧基(或氟)形成三中心氢键, 因此也曾被用于构建折叠体[22,34]. HO-O, HF-F和HF-O在氯仿和DMSO中的Δ(δH)-1值处在0.43~0.49之间, 说明它们具有近平面的构象及三中心氢键的形成(表7). 此外, 我们还合成了含脲基的模型化合物UEs-Es(Scheme 3), 其在氯仿和DMSO中的Δ(δγH)-1值为0.44(表S2, 见本文支持信息), 表明N—H…O氢键具有较好的构象控制效果, 它的两个酰胺NH可以分别与两个甲酯基团中的羰基形成二中心氢键.
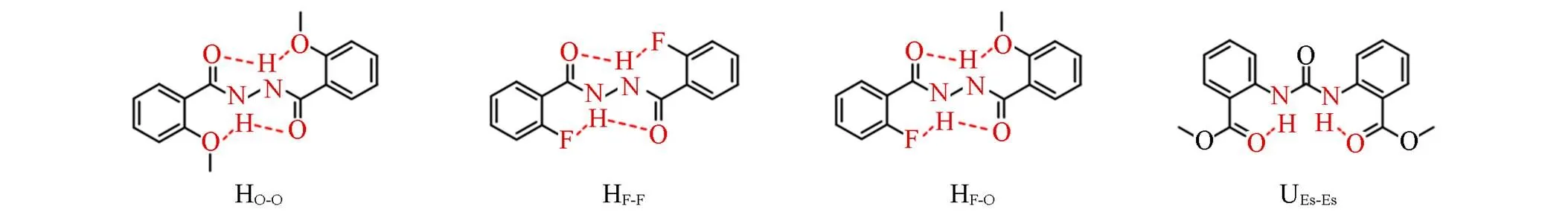
Scheme 3Chemical structures of other aromatic compounds that can form intramolecular hydrogen bonds
HO-O: 2-methoxybenzoic acid'-(2-methoxybenzoyl)hydrazide; HF-F: 2-fluoro-'-(2-fluorobenzoyl)benzohydrazide;HF-O: 2-fluoro-'-(2-methoxybenzoyl)benzohydrazide; UEs-Es: 1,3-bis[2-(methoxylcarbonyl)phenyl]urea.
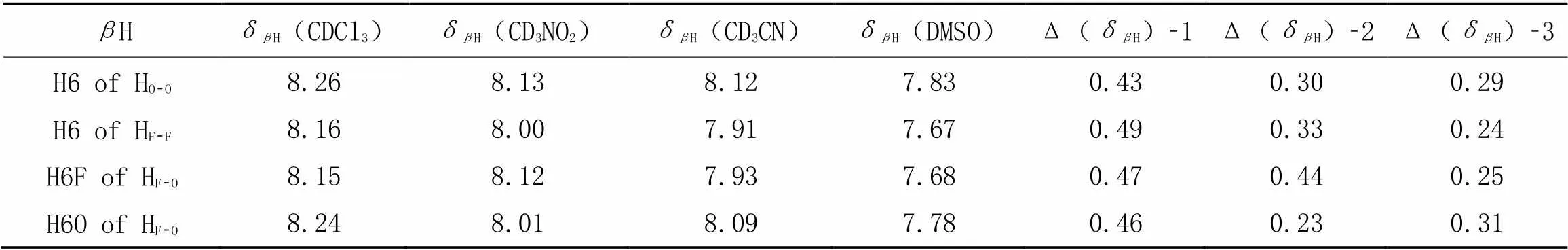
Table 7 Chemical shifts ofβH of HO-O, HF-F and HF-O in CDCl3 and DMSO⁃d6, and their solvent⁃related changes*
*Δ(δH)-1=δH(CDCl3)-δH(DMSO), Δ(δH)-2=δH(CD3NO2)-δH(DMSO), Δ(δH)-3=δH(CD3CN)-δH(DMSO).
2.5 三中心氢键和二中心氢键模型化合物的对比
对比分析表2, 表4和表6中三中心氢键和二中心氢键模型化合物的Δ(δH)-1的值可以发现如下规律: 对于具有相同苯胺部分的一组-芳基芳酰胺化合物, 二中心氢键模型化合物的Δ(δH)是最大的. 例如: 对于含有2-甲氧基苯胺的4个-芳基芳酰胺化合物, 二中心氢键模型化合物CPh-O的Δ(δH)-1最大(0.74). 依据这些数据, 我们推测三中心氢键模型化合物的两个氢键受体之间是竞争关系, 其中一个受体与酰胺NH形成分子内氢键后, 不利于酰胺NH再与另一个受体形成分子内氢键. 例如: 由于氟原子的竞争, CF-O的N—H…O分子内氢键在某种程度上比CPh-O的分子内氢键弱.
2.6 三中心氢键模型化合物Δ(δβH)和Δ(δγH)的比较
表8给出9个三中心氢键模型化合物在氯仿和DMSO中Δ(δH)和Δ(δH)的分析结果. 可见, CF-F, CF-O和CO-F这3个模型化合物的Δ(δH)和Δ(δH)值都比较大, 说明CF-F, CF-O和CO-F这3种结构单元在酰胺基团的左右两侧都能获得很好的构象控制效果. 此外, 分析了这些化合物在乙腈和DMSO中Δ(δH)和 Δ(δH)的数值(表S3, 见本文支持信息), 发现了与表8一致的规律. 虽然这3种结构单元在构建折叠体时展示出很好的构象控制效果, 但相关报道并不多. 例如, 曾华强等[35]利用CF-F基元制备了一个大环结构, 利用CF-O和CO-F基元限制了两个折叠体低聚物的局部构象, 黎占亭等[19,34,36~39]利用CF-O和CF-F基元制备了多个折叠体低聚物, 晶体学研究证明了这些结构的平面性和三中心氢键的存在, 并认为N—H…F氢键比N—H…O氢键具有更加平面的结构[40].

Table 8 Solvent-related changes in chemical shifts of βH and γH of aromatic amides in CDCl3 and DMSO-d6*
* Δ(δH)=δH(CDCl3)-δH(DMSO); Δ(δH)=δH(CDCl3)-δH(DMSO).
3 结 论
应用Δ(δH)方法分析了一系列芳酰胺的一维核磁共振氢谱, 利用Δ(δH)和Δ(δH)的值能够分别判断这两个质子是否处于羰基的去屏蔽区, 进而更准确地分析-芳基芳酰胺的构象以及三中心氢键. 基于1H NMR分析结果, CF-F, CF-O和CO-F这3种-芳基芳酰胺结构在溶液中具有良好的平面构象, 因此认为它们在构建折叠体方面的潜力有待进一步挖掘. 此外, 基于多组核磁数据的对比, 我们发现当NH与第二个氢键受体形成氢键时, 其与第一个氢键受体之间的氢键就被削弱了, 换言之, 在芳酰胺形成三中心氢键时, 两个氢键受体争相与NH形成氢键并取得了某种平衡.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220084.
[1] Jeffrey G. A., Saenger W.,, Springer, Heidelberg, 1991
[2] Kuhn B., Mohr P., Stahl M.,...,2010,(6), 2601—2611
[3] Huc I.,....,2004,(1), 17—29
[4] Zhang D. W., Zhao X., Hou J. L., Li Z. T.,..,2012,(10), 5271—5316
[5] Zhang D. W., Wang W. K., Li, Z. T.,..,2015,(1), 233—251
[6] Jeffrey G. A., Mitra J.,....,1984,(19), 5546—5553
[7] Rozas I., Alkorta I., Elguero J.,...,1998,(48), 9925—9932
[8] Kim S. G., Kim K. H., Kim Y. K., Shin S. K., Ahn K. H.,....,2003,(45), 13819—13824
[9] Taylor R., Kennard O., Versichel W.,....,1984,(1), 244—248
[10] Garric J., Léger J. M. Huc I.,..,..,2005,(13), 1954—1958
[11] Lu Y. X., Shi Z. M., Li, Z. T., Guan Z.,..,2010,(47), 9019—9021
[12] Xin P., Zhu P., Su P., Hou J. L., Li Z. T.,....,2014,(38), 13078—13081
[13] Zhu J., Parra R. D., Zeng H., Skrzypczak⁃Jankun E., Zeng X. C., Gong B.,....,2000,(17), 4219—4220
[14] Parra R. D., Zeng H., Zhu J., Zheng C., Zeng X. C., Gong B.,...,2001,(20), 4352—4357
[15] Gong B., Zeng H., Zhu J., Yuan L., Han Y., Cheng S., Furukawa M., Parra R. D., Kovalevsky A. Y., Mills J. L., Skrzypczak⁃Jankun E., Martinovic S., Smith R. D., Zheng C., Szyperski T., Zeng X. C.,.......,2002,(18), 11583—11588
[16] Manjunatha Reddy G. N., Vasantha Kumar M. V., Guru Row T. N., Suryaprakash N.,....,2010,(40), 13232—13237
[17] Hamuro Y., Geib S. J., Hamilton A. D.,.....,1994,(4), 446—448
[18] Gong B.,...,2001,(20), 4336—4342
[19] Li C., Ren S. F., Hou J. L., Yi H. P., Zhu S. Z., Jiang X. K., Li Z. T.,....,2005,(35), 5725—5729
[20] Ong W. Q., Zhao H., Fang X., Woen S., Zhou F., Yap W., Su H., Li S. F. Y., Zeng H.,..,2011,(12), 3194—3197
[21] You L. Y., Chen S. G., Zhao X., Liu Y., Lan W. X., Zhang Y., Lu H. J., Cao C. Y., Li Z. T.,....,2012,(7), 1657—1661
[22] Hou J. L., Shao X. B., Chen G. J., Zhou Y. X., Jiang X. K., Li, Z. T.,....,2004,(39), 12386—12394
[23] Zhang D. W., Wang H., Li Z. T.,, 2020,(11), 1665—1679(张丹维, 王辉, 黎占亭. 化学进展, 2020,(11), 1665—1679)
[24] Gan Q., Li F., Li G., Kauffmann B., Xiang J., Huc I., Jiang H.,..,2010,(2), 297—299
[25] Gan Q., Bao C., Kauffmann B., Grélard A., Xiang J., Liu S., Huc I., Jiang H.,....,2008,(9), 1715—1718
[26] Zhu J., Wang X. Z., Chen Y. Q., Jiang X. K., Chen X. Z., Li Z. T.,...,2004,(19), 6221—6227
[27] Ernst J. T., Becerril J., Park H. S., Yin H., Hamilton A. D.,....,2003,(5), 535—539
[28] Shi Z. M., Song Y., Lu F., Zhou T. Y., Zhao X., Zhang W. K., Li Z. T.,,2013,(1), 51—61(施朱明, 宋宇, 陆方, 周天佑, 赵新, 张文科, 黎占亭.化学学报,2013,(1), 51—61)
[29] Zhang J., Chen P., Yuan B., Ji W., Cheng Z., Qiu X.,,2013,(6158), 611—614
[30] Mönig H., Amirjalayer S., Timmer A., Hu Z., Liu L., Díaz Arado O., Cnudde M., Strassert C. A., Ji W., Rohlfing M., Fuchs H.,..,2018,(5), 371—375
[31] Abraham R. J., Mobli M., Smith R. J.,...,2003,(1), 26—36
[32] Abraham R. J., Griffiths L., Perez M.,...,2014,(7), 395—408
[33] Min J., Wang C., Wang L.,....,2021,(23), 13284—13291
[34] Zhu Y. Y., Wu J., Li C., Zhu J., Hou J. L., Li C. Z., Jiang X. K., Li Z. T.,..,2007,(8), 1490—1496
[35] Liu Y., Shen J., Sun C., Ren C., Zeng H.,....,2015,(37), 12055—12063
[36] Liu C. Z., Koppireddi S., Wang H., Zhang D. W., Li, Z. T.,....,2019,(1), 226—230
[37] Liu C. Z., Koppireddi S., Wang H., Zhang D. W., Li Z. T.,...,2019,(5), 953—956
[38] Koppireddi, S., Liu C. Z., Wang H., Zhang D. W., Li Z. T.,,2019,(16), 2626—2630
[39] Li C., Zhu Y. Y., Yi H. P., Li C. Z., Jiang X. K., Li Z. T., Yu Y. H.,..,2007,(35), 9990—9998
[40] Zhu Y. Y., Li C., Li G. Y., Jiang, X. K., Li Z. T.,...,2008,(5), 1745—1751
1H NMR Study on the Conformation of Aromatic Amides Limited by Three-center Hydrogen Bonds
MINJing, WANGLiyan*
(,,,130012,)
We designed and synthesized nine-aryl aromatic amides that could form three-center hydrogen bonds, and six-aryl aromatic amides that could form two-center hydrogen bonds. Based on their one-dimensional1H nuclear magnetic resonance(NMR) spectra in chloroform and DMSO, the de-shielding effects of carbonyl groups onH andH were analyzed separately. The conformational restriction of aromatic amides by three-center hydrogen bonds was evaluated by combining Δ(δH) and Δ(δH). It was disclosed that three aromatic amides [2-fluoro--(2-methoxyphenyl)benzamide,-(2-fluorophenyl)-2-methoxybenzamide and 2-fluoro--(2-fluorophenyl)benzamide] display excellent planarization of molecular comformations at both sides of their carbonyl groups, which would have greater potential in constructing new foldamers. Furthermore, it was inferred that, when an NH group forms hydrogen bond with the second hydrogen-bond acceptor, the hydrogen bond between the NH group and the first hydrogen-bond acceptor would be weakened. In other words, for an aromatic amide with a three-center hydrogen bond, two hydrogen-bond acceptors contend with each other for the NH group, resulting in a balanced situation.
Three-center hydrogen bond; Aromatic amide; Conformation; Foldamer
O657
A
10.7503/cjcu20220084
2022-02-12
2022-03-17.
简介: 王力彦, 男, 博士, 教授, 主要从事聚合物自组装与非共价键方面的研究. E-mail: wangliyan@vip.sina.com
国家自然科学基金(批准号: 22193020, 22193024, 21827805)资助.
Supported by the National Natural Science Foundation of China(Nos.22193020, 22193024, 21827805).
(Ed.: W, K, M)
