Ag, Zn, Pd掺杂对铜基催化剂草酸二甲酯选择性加氢反应的影响
2022-07-18宋有为安江伟王征王旭慧权燕红任军赵金仙
宋有为, 安江伟, 王征, 王旭慧, 权燕红, 任军, 赵金仙
Ag, Zn, Pd掺杂对铜基催化剂草酸二甲酯选择性加氢反应的影响
宋有为, 安江伟, 王征, 王旭慧, 权燕红, 任军, 赵金仙
(太原理工大学省部共建煤基能源清洁高效利用国家重点实验室, 太原 030024)
采用密度泛函理论方法, 构建了Ag, Zn, Pd原子掺杂的Cu(111)和Cu2O(111)活性晶面, 探讨了不同金属掺杂对Cu(111)和Cu2O(111)催化剂的草酸二甲酯(DMO)加氢反应活性和选择性的影响. 研究结果显示, 掺杂Zn可有效阻止乙醇酸甲酯(MG)进一步加氢, 提高MG的选择性, Ag助剂可以有效提高加氢活性; 而Pd助剂的添加使MG的生成能垒增高, 降低了MG的选择性. Ag-Cu(111)表面具有适宜的带中心, 生成CH3OOCCH2OH的活性最高. 在Ag, Zn, Pd原子掺杂的Cu2O(111)表面, Ag-Cu2O(111)能带带隙小、 价带强度高, 在DMO加氢反应中具有最佳的催化活性. 基于上述结果, 提出铜基催化剂结构调变和性能调控的理论方法, 为高效催化剂的设计提供可靠的理论指导.
草酸二甲酯; 选择性加氢; 铜基催化剂; 金属掺杂
开发高附加值的下游化学品是煤炭清洁高效利用的重要研究方向[1,2]. 乙醇酸甲酯(MG)是一种用途广泛的精细化学品, 是重要的化工中间体和高品质溶剂[3]. 草酸二甲酯(DMO)选择性加氢反应合成MG, 具有原子利用率高(64%)、 生产工艺绿色环保的优势, 是一条非常有发展前途的新型煤化工产品工艺路线[4,5]. 对于DMO加氢合成MG反应, 铜基催化剂价格低廉、 C=O键加氢活性较高, 引起了国内外研究者的广泛关注. 实验研究[6,7]与密度泛函理论(DFT)计算[8]结果表明, 铜基催化剂中Cu0与Cu+双活性位及其协同催化作用主导DMO加氢反应的性能, Cu0与Cu+的稳定形成与平衡共存是达到优异催化性能的前提条件. 然而, 在反应过程中, 铜纳米颗粒易于迁移聚集, 且铜物种价态不稳定, 导致活性比表面积降低及活性物种Cu0与Cu+的比例失衡, 最终影响催化活性及稳定性[9]. 在金属催化剂中掺杂助剂进而改变其组成和结构, 是调控加氢反应性能的有效途径[10,11]. 分析显示, 金属银、 锌和钯与铜晶格参数接近, 都具有面心立方(FCC)结构, 有相似的电子结构和物理化学性质. 在DMO加氢反应中, Wang等[3]将Ag助剂加入到Cu/SiO2催化剂体系中, 发现在较高的液时空速下, MG能保持较高的收率. Zhang等[12]通过改变Cu-Pd/SiO2催化剂中Pd的含量来控制Cu0/Cu+的比例, 发现添加适量的Pd可以显著提高催化剂的活性, DMO转化率达到100%, 乙二醇(EG)选择性达到96%, 延长了催化剂的使用寿命(300 h). 该研究认为, 催化剂性能的提升可归因于通过氢溢流的产生调整了Cu0/Cu+比例, 并且通过促进与铜离子接触的中间体的释放抑制了铜的烧结. Wang等[13]研究表明, 在铜催化剂中掺入适量的Zn能够提高表面Cu+含量, 促进Cu0颗粒分散, 提高抗烧结性能, DMO转化率为99.5%, 对乙二醇的选择性为91.5%.
在DMO加氢体系中, 有关铜基催化剂掺杂第二金属的报道很多, 但对掺杂金属的选择尚处于经 验性的尝试阶段, 缺乏理论指导. 本文采用DFT计算方法探讨了掺杂Ag, Zn, Pd原子的Cu(111)和Cu2O(111)晶面上DMO加氢反应机理, 讨论3种金属掺杂铜催化剂双活性位结构与性能之间的关系, 考察了基元反应进行的难易程度, 揭示了催化剂表面结构与反应性能之间的关系, 进而合理精确地预测了催化剂的DMO加氢反应性能, 提出了从电子-分子水平上可供参考或借鉴的催化剂结构模型, 进而指导催化剂的设计和制备.
1 计算方法和模型
所有的DFT计算均采用Materials Studio 5.5中Dmol3模块进行[14]. 交换关联泛函采用广义梯度近 似GGA-PBE方法[15,16], 价电子波函数采用双数值基加极化函数(DNP)展开, 金属原子采用有效核电 势(ECP)[17], 其它原子采用全电子(All electron)基组. 布里渊区积分Monkhorst-Pack网格参数设为 4×4×1[18]. 使用完全线性同步/二次线性同步(LST/QST)方法进行过渡态计算[19], 并进行频率分析. Hirschfeld电荷分析方法用于计算电荷转移[20]. 数字积分采用程序中的Medium水平, 能量、 受力和 位移收敛标准分别是5.44×10‒4eV, 1.09 eV/nm和5×10‒4nm, 自洽场(SCF)密度收敛设置为2.72× 10‒4eV, 拖尾效应(Smearing)设置为0.136×10‒3eV.
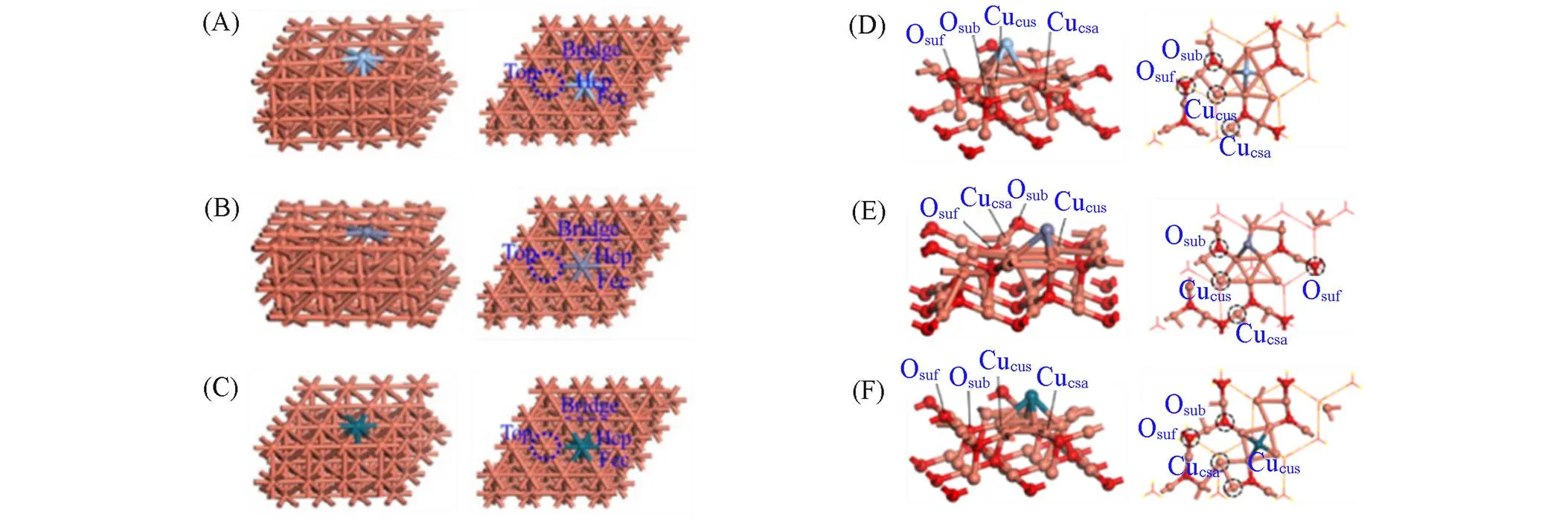
Fig.1 Side views(left) and top views(right) of Ag⁃Cu(111)(A), Zn⁃Cu(111)(B), Pd⁃Cu(111)(C), Ag⁃Cu2O(111)(D), Zn⁃Cu2O(111)(E) and Pd⁃Cu2O(111)(F) models
如图1(A)~(C)所示, 采用3层(4×4)超胞来模拟Cu(111)表面, 弛豫顶层原子, 固定底部两层原子, 用Ag, Zn, Pd原子分别取代Cu(111)表面的1个Cu原子, 模拟Ag, Zn, Pd掺杂Cu0催化剂, 标记为-Cu(111)催化剂(=Ag, Zn或Pd). 以上催化剂模型均包含4种吸附位点: 顶位、 桥位、 Fcc和Hcp. 所有表面模型中真空层为2 nm.
如图1(D)~(F)所示, 采用6层(2×2)超胞来模拟Cu2O(111)表面, 弛豫上面3层原子, 固定底部 3层原子, 将表面的一个氧原子分别用1个Ag, Zn, Pd原子代替, 构建-Cu2O(111)模型(=Ag, Zn或Pd).-Cu2O(111)模型均包括4种吸附位点: Cucus, Cucsa, Osuf和Osub, 其中Cucus和Cucsa位点分别是配位不饱和Cu和饱和Cu原子, Osuf和Osub位点分别是表面O和次表面O原子.
Zhao等[21]利用助剂Zn掺杂CeO2(111)表面的模型, 研究了助剂Zn对CO2和CH4在CeO2催化剂上的C—C偶联反应的影响, 通过用1个Zn原子取代1个Ce原子构建了助剂Zn掺杂CeO2(111), 这与构建Ag-Cu2O(111), Zn-Cu2O(111)和Pd-Cu2O(111)催化剂的表面重构方法相似, 且均包括4种吸附位点: Cucus, Cucsa, Osuf和Osub, 其中Cucus和Cucsa位点分别是配位不饱和Cu和饱和Cu原子, Osuf和Osub位点分别是表面O和次表面O原子. 在所有表面模型中, 设置一个2 nm的真空层.
2 结果与讨论
2.1 M-Cu(111)催化剂
2.1.1物种的吸附反应物种在催化剂表面的吸附能(ads, kJ/mol)的计算公式为
ads(A) =(Cu+A)-A/Cu(1)
式中:A,Cu和A/Cu(kJ/mol)分别表示吸附物、 铜基催化剂和含吸附物的铜基催化剂的能量. 吸附能为正值表示放热, 负值表示吸热.
图2和表1给出了反应物H2在M-Cu(111)(M=Ag, Zn或Pd)表面的最稳定吸附位及相应的吸附能. 与H2分子在Cu(111)表面的解离吸附类似[8], 在M-Cu(111)表面, 反应物H2平行侧向吸附于M-Cu(M=Ag, Zn, Pd)桥位, 并发生解离吸附. 与Cu(111)表面相比, H物种在Ag-Cu(111), Zn-Cu(111)和Pd-Cu(111)表面的吸附强度相差不大.

Fig.2 Stable configurations of H2 dissociative adsorption on Ag⁃Cu(111)(A), Zn⁃Cu(111)(B) and Pd⁃Cu(111)(C) catalysts

Table 1 Adsorption sites and adsorption energies of all species involved in DMO hydrogenation to MG on Cu(111), Ag-Cu(111), Zn-Cu(111) and Pd-Cu(111) surfaces
图3列出了反应物DMO、 可能的中间体和产物在M-Cu(111)(M=Ag, Zn, Pd)催化剂表面上的最 稳定吸附构型, 相应的吸附位点和吸附能如表1所示. 反应物DMO分子在M-Cu(111)表面吸附较弱, 与催化剂之间并未形成稳定的化学键, 然而吸附能均高于其在Cu(111)表面的吸附能(46.0 kJ/mol). 与Cu(111)表面相比, H物种在Ag-Cu(111)和Zn-Cu(111)表面的吸附强度减弱. 关键的中间体CH3OOCCO[22]在Pd-Cu(111)表面的吸附强度(199.6 kJ/mol)显著增强, 在Ag-Cu(111)和Zn-Cu(111)表面吸附能变化不大. CH3OOCCO在Pd-Cu(111)表面通过C原子吸附于Pd位点, 而在Ag-Cu(111)和 Zn-Cu(111)表面通过C原子吸附于Cu位点. CH3O物种在Ag-Cu(111)和Pd-Cu(111)表面的吸附能较Cu(111)表面有所降低, 而在Zn-Cu(111)表面的吸附能变化不大. CH3OOCCOH和CH3OOCCHOH中间体在Pd-Cu(111)的吸附能明显高于Ag-Cu(111)和Zn-Cu(111)表面, 表明Pd助剂的添加可以稳定CH3OOCCOH和CH3OOCCHOH中间体. 产物MG分子在-Cu(111)(=Ag, Zn, Pd)表面均为弱吸附. 与Cu(111)相比,-Cu0催化剂表面上MG的吸附能均有所降低, 表明产物MG容易从催化剂表面脱离.
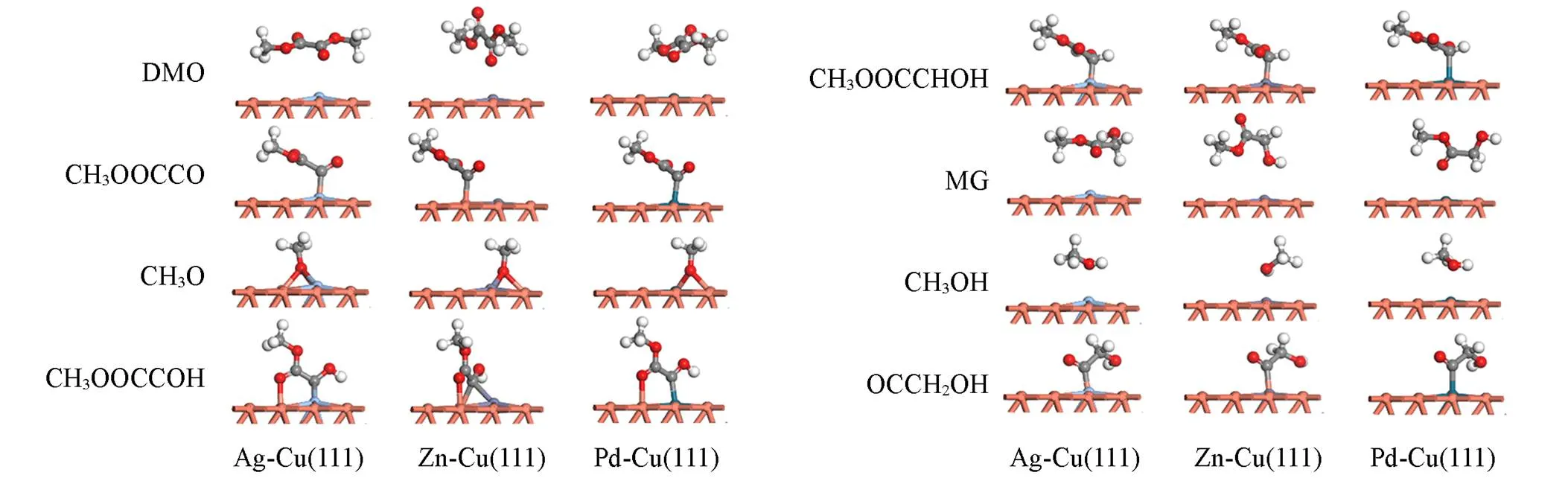
Fig.3 Most stable adsorption configurations of species involved in DMO hydrogenation to MG over M⁃Cu(111) surface
在DMO加氢反应中, MG可以解离生成OCCH2OH物种. 从表1中可以看出, 在-Cu(111)催化剂表面, OCCH2OH物种的吸附能遵循以下顺序: Pd-Cu(111)>Cu(111)>Ag-Cu(111)>Zn-Cu(111), 表明Pd助剂显著增强了OCCH2OH物种的稳定性. 同时, 副产物CH3OH分子在Ag-Cu(111), Zn-Cu(111)和Pd-Cu(111)催化剂表面的吸附能均小于45 kJ/mol, 因此, CH3OH分子在-Cu(111)催化剂表面为物理吸附, 可以从催化剂表面及时脱附.
2.1.2DMO加氢机理根据文献[23]报道, 总结了Cu基催化剂表面DMO加氢的可能的反应路径, 如 表2所示. 各基元反应的能垒(a, kJ/mol)和反应热(Δ, kJ/mol)的计算公式如下:
a=TS-IS(2)
Δ=FS-IS(3)
式中:TS,IS和FS分别为基元反应中过渡态、 初态和终态的总能量.
不同金属掺杂Cu0催化剂表面H2解离吸附构型分别如图2所示. 从图2和表2可以看出, H2分子在Ag-Cu(111), Zn-Cu(111)和Pd-Cu(111)催化剂表面直接解离为活性H原子. DMO分子在Ag-Cu(111)催化剂表面解离生成CH3OOCC*O和CH3O*, 需要克服224.3 kJ/mol能垒, 与其在Cu(111)表面所需要的能量(220.2 kJ/mol)相差无几; 在Pd-Cu(111)表面, DMO解离所需要克服的能垒为327.2 kJ/mol; 而在Zn-Cu(111)表面, DMO解离的活化能仅为158.5 kJ/mol. 因此, Zn助剂的添加促进了Cu(111)催化剂表面DMO的解离.
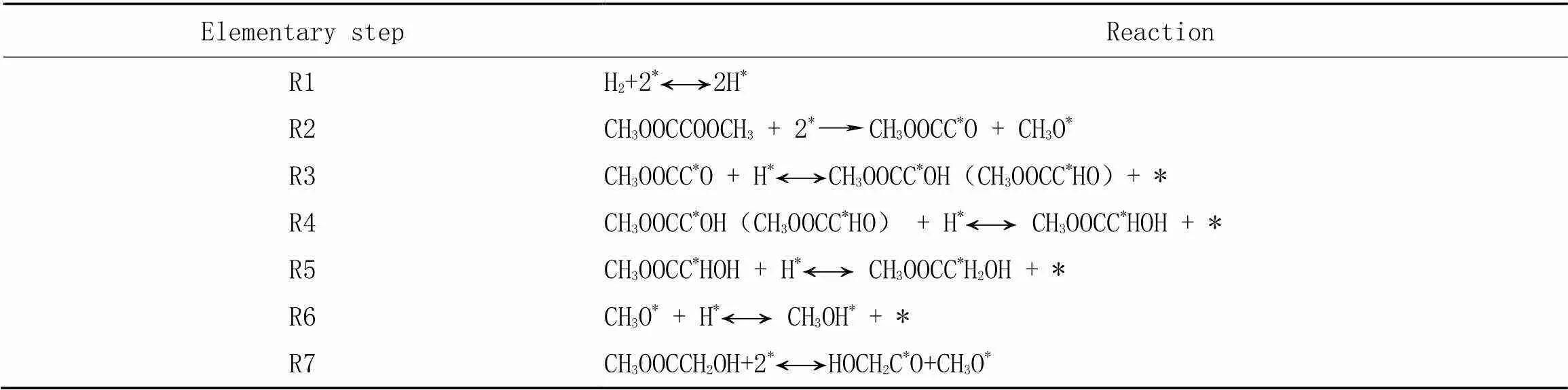
Table 2 Mechanism of DMO hydrogenation over Cu catalyst#
# * Stands for active sites.
由表2可以看出, CH3OOCC*O物种在M-Cu(111)(M=Ag, Zn, Pd)催化剂表面逐步加氢生成CH3OOCC*OH(或CH3OOCC*HO), CH3OOCC*HOH和CH3OOCC*H2OH(表2中反应R3~R5). 研究表明, 在关键中间物种CH3OOCCO[24]加氢生成MG的过程中, Ag, Zn, Pd金属的掺杂使得CH3OOCCO加氢路径发生改变. 由于CH3OOCCHO中间体在Ag-Cu(111), Zn-Cu(111)和Pd-Cu(111)催化剂表面均不能稳定存在, 且加氢生成CH3OOCCHO活化能较生成CH3OOCCOH活化能更高, 因此, CH3OOCCOH中间体优先生成, 即H原子首先与酰基物种末端C=O键中的O原子结合[22]. 随后, H原子被依次加到酰基物种末端C=O键中的C原子上, 得到CH3OOCCHOH和CH3OOCCH2OH.
从表1和表3中可以看出, CH3OOCC*O和CH3OOCC*OH物种在-Cu(111)表面的吸附能均大于其加氢反应的活化能[25,26], 因此, CH3OOCC*O和CH3OOCC*OH物种优先发生加氢反应, 但是加氢能垒均高于纯Cu(111)催化剂表面(60.1和23.7 kJ/mol). 对于CH3OOCC*HOH物种, 在Ag-Cu(111)和Zn-Cu(111)催化剂表面吸附能大于加氢活化能, 优先发生加氢反应(表3中反应R5); 而在Pd-Cu(111)表面则相反, 加氢反应的活化能大于其吸附能, CH3OOCC*HOH物种优先发生脱附.
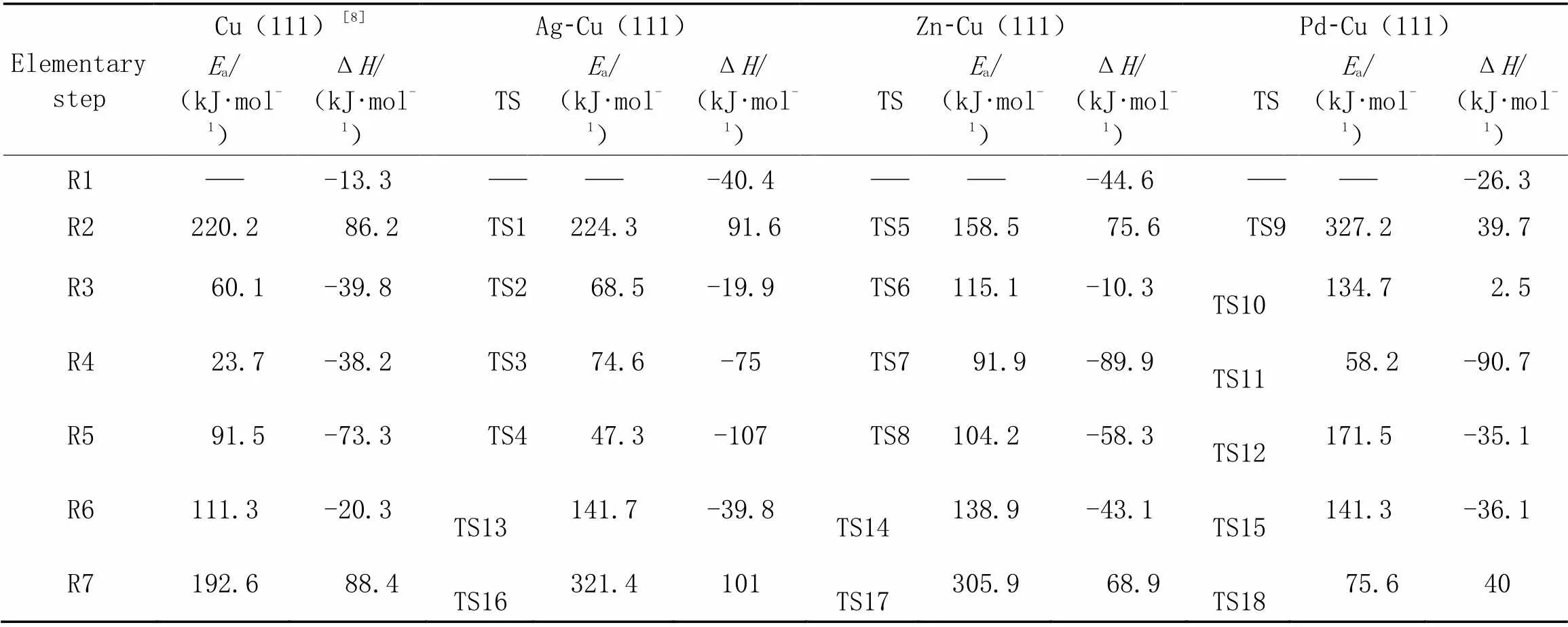
Table 3 Activation energies(Ea) and reaction energies(ΔH) for elementary reactions(R1—R5) of DMO hydrogenation to MG and side reactions(R6, R7) on Cu(111) and M-Cu(111) surfaces
2.2 M-Cu2O(111)催化剂
2.2.1物种的吸附图4和图5分别给出了DMO选择性加氢生成MG过程中涉及到的反应物、 可能的中间体以及产物在M-Cu2O(111)表面的稳定吸附构型, 相应的稳定吸附位和吸附能分别列于表4.
与Cu(111)表面相比, H物种在Ag-Cu(111), Zn-Cu(111)和Pd-Cu(111)表面的吸附强度相差不大. 从图4可以看出, H2在Zn-Cu2O(111)表面直接解离吸附, 不需要克服能垒即产生活性氢原子; 而在Ag-Cu2O(111)和Pd-Cu2O(111)表面则为弱吸附, H2分子侧向吸附于Cucus位点. 在M-Cu2O(111)表面则通过O原子稳定吸附于Cucus位点, 稳定吸附构型为分子形式吸附, 吸附能相差不大, 且略低于其在Cu2O(111)表面的吸附能(101.3 kJ/mol).
与M-Cu(111)(M=Ag, Zn, Pd)催化剂不同的是, 在M-Cu2O(111)表面, 生成CH3OOCCOH的能垒明显高于CH3OOCCHO物种, 因此, CH3OOCCO物种加氢优先生成CH3OOCCHO中间体[27]. 随后, H原子被依次加到酰基物种末端C=O键中的O原子和C原子上, 逐步得到CH3OOCCHOH和CH3OOCCH2OH. 结果表明, Ag, Zn, Pd助剂的加入均显著提高了CH3OOCCHO和CH3OOCCHOH的吸附能. MG在M-Cu2O催化剂表面的吸附能则高于其在Cu2O表面的吸附能, 因此, 与Cu2O(111)相比, M-Cu2O(M=Ag, Zn, Pd)催化剂表面MG分子脱附比较困难.
与Cu(111)表面不同的是, 副产物CH3OH分子均可以稳定吸附在Cu2O(111)和M-Cu2O(111)催化剂表面, 其吸附能均大于85 kJ/mol, 说明副产物CH3OH在M-Cu2O(111)催化剂表面脱附比较困难.

Fig.4 Initial states, transition states(TS), and final states of H2 dissociation over Ag⁃Cu2O(111)(A), Pd⁃Cu2O(111)(B) and Zn⁃Cu2O(111)(C) surfaces

Fig.5 Most stable adsorption configurations of the species involved in DMO hydrogenation to MG over M⁃Cu2O(111) surfaces
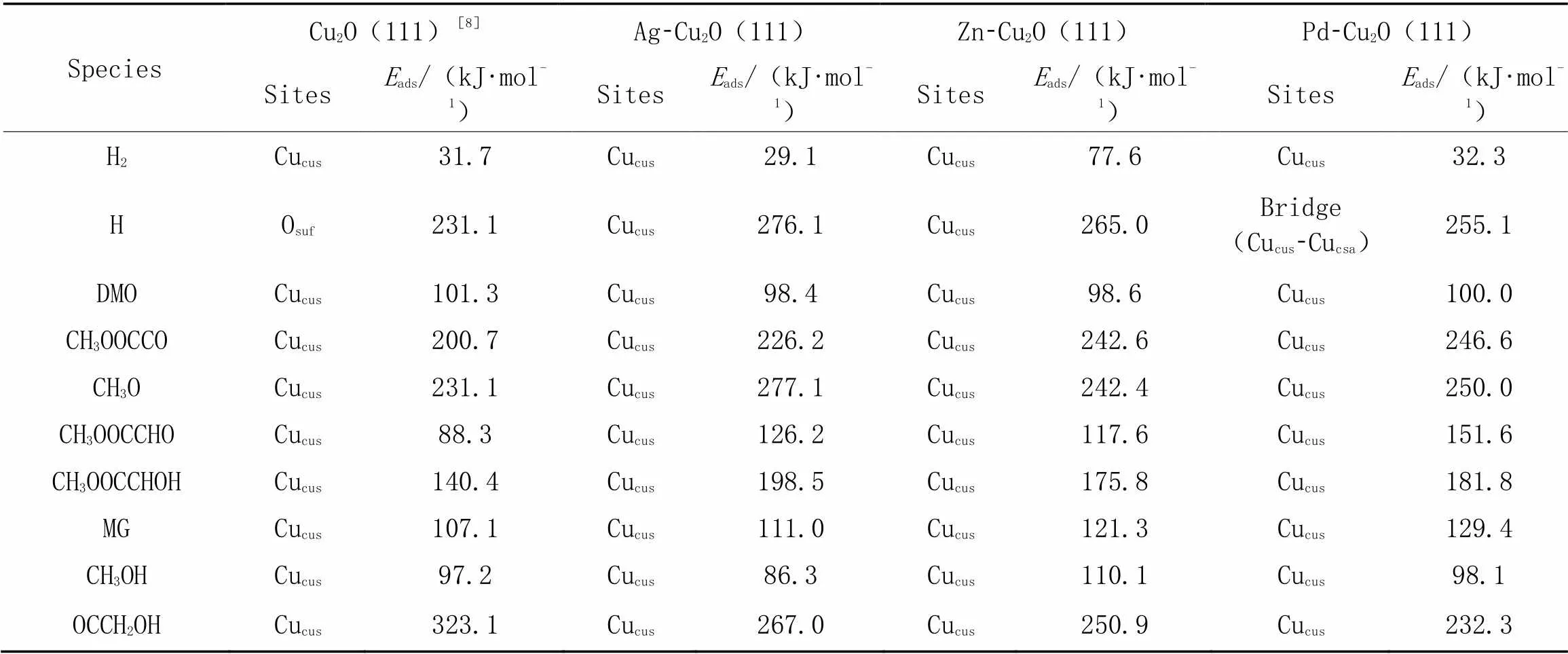
Table 4 Adsorption sites and adsorption energies(Eads) of all species involved in DMO hydrogenation to MG on Cu2O(111) and M⁃Cu2O(111) surfaces
2.2.2DMO加氢机理与M-Cu(111)催化剂类似, 平行吸附于Zn-Cu2O(111)表面Cucsa-Cucas桥位的H2分子直接解离, 而在Ag-Cu2O(111)和Pd-Cu2O(111)催化剂表面, H2分别经过渡态TS37和TS38解离产生氢原子(图4), 但是其活化能均远低于Cu2O(111)表面所需的能量(299 kJ/mol)(表5). 因此, 助剂Ag, Zn和Pd的添加促进了H2在Cu2O(111)表面的解离. Ag, Zn和Pd助剂的添加均使得DMO在Cu2O(111)表面的解离能垒增加(表5).
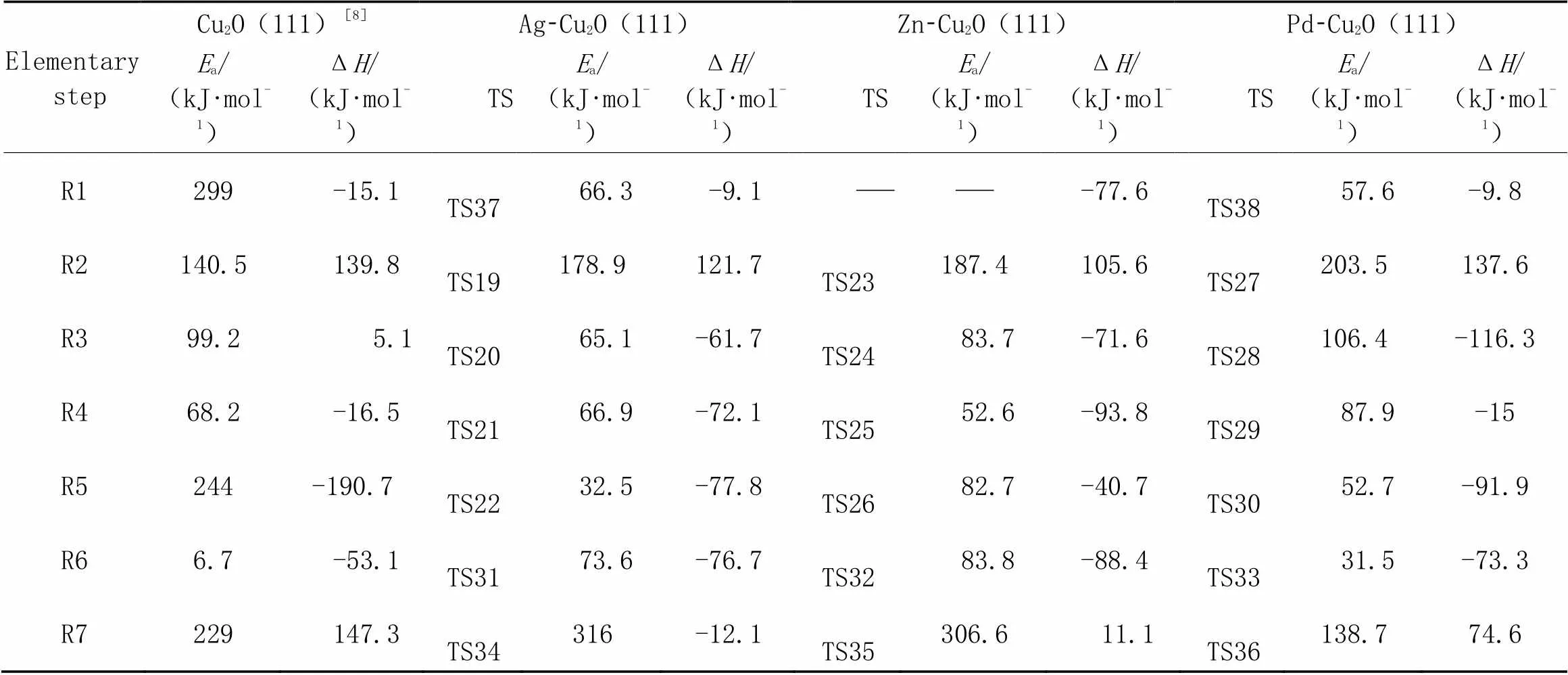
Table 5 Activation energies(Ea) and reaction energies(ΔH) for elementary reactions(R1—R5) of DMO hydrogenation to MG and side reactions(R6, R7) on Cu2O(111) and M-Cu2O(111) surfaces
在-Cu2O(111)催化剂表面, CH3OOCC*O, CH3OOCC*HO和CH3OOCC*HOH物种的加氢能垒(表5)明显小于其吸附能(表4)[25], 因此均优先发生加氢反应. 对于加氢反应R3和R4, 助剂Ag和Zn显著降低了Cu2O催化剂的加氢能垒, 而Pb则使反应能垒升高. 在CH3OOCC*HOH加氢反应(R5)中, 助剂Ag, Zn和Pd均大幅度地降低了加氢能垒, 极大地提高了Cu2O催化剂的加氢性能. Ag, Zn, Pd助剂使得H, CH3OOCCO, CH3OOCCHO和CH3OOCCHOH物种的吸附能增强, 改善了Cu+催化剂表面的加氢活性. 综合分析认为, Ag-Cu2O(111)在加氢过程中催化性能最佳.
2.3 副反应
从表3和表5中可以看出, 与Cu0和Cu+催化剂相比, Ag, Zn和Pd助剂的添加均提高了副产物CH3OH的生成能垒. 然而, CH3OH的生成能垒均低于CH3O*物种的脱附能(表1和表4), 表明在Cu0和Cu+催化剂表面, CH3O*物种更倾向于加氢生成CH3OH. Ag和Zn助剂的添加使得MG的解离活化能远高于其在纯Cu0和Cu+催化剂的解离能垒. 因此, Ag和Zn助剂可以有效阻止MG的进一步解离加氢, 提高了MG的选择性. 而Pd助剂则降低了MG的解离能垒, MG在Pd-Cu2O(111)催化剂表面的解离能垒与其相应的吸附能接近, 使得MG的脱附和解离形成竞争反应, 导致MG选择性降低.
2.4 讨 论
2.4.1势能面和反应机理从图6可以看出, 在-Cu(111)(=Ag, Zn, Pd)和Cu(111)表面, DMO加氢生成MG的速控步骤均为DMO解离为CH3OOCCO*, Ag, Zn或Pd助剂的添加没有改变该反应的速控步骤. 与Cu(111)相比, Pd助剂提高了DMO解离能垒, Ag-Cu(111)表面DMO解离能垒变化不大, 而 Zn显著降低了催化剂表面DMO的解离能垒, 表明Zn-Cu(111)催化剂表面发生DMO加氢反应是热力学上最有利的[28].

Fig.7 Energy barrier of DMO hydrogenation over Cu2O(111) and M⁃Cu2O(111) catalysts

Fig.6 Energy barrier of DMO hydrogenation over Cu(111) and M⁃Cu(111) catalysts
在Cu2O表面, Ag, Zn或Pd助剂的添加使得DMO加氢反应的速控步骤由H2解离变为DMO的解离(图7). 4种Cu+催化剂上DMO加氢反应中速控步骤的能垒从低到高依次为Ag-Cu2O(111)(178.9 kJ/mol) < Zn-Cu2O(111)(187.4 kJ/mol) < Pd-Cu2O(111)(203.5 kJ/mol) < Cu2O(111)(299.0 kJ/mol). 因此, Ag助剂可以有效提高Cu2O(111)催化剂的DMO加氢活性.
2.4.2Hirschfeld电荷分析和带中心分析为了深入探究Zn和Ag助剂对Cu(111)和Cu2O(111)影响的根本原因, 对催化剂及反应物体系进行了Hirschfeld电荷分析.
从表6中可以看出, Cu(111), Ag-Cu(111), Zn-Cu(111)和Pd-Cu(111)催化剂表面与DMO分子之间转移电荷量分别为0.0194|e|, -0.0942|e|, -0.0038|e|和-0.1498|e|, MG分子带有的电荷量分别为 -0.0630|e|, -0.0851|e|, -0.0702|e|和-0.0283|e|. 图8为Cu(111)和M-Cu(111)催化剂表面单个Cu原子的带偏态密度图. Zn-Cu(111), Ag-Cu(111), Cu(111)和Pd-Cu(111)催化剂表面上单个Cu原子的带中心分别为-2.50, -2.45, -2.44和-2.41 eV. 相关文献[29,30]报道, 金属催化剂的电子结构对其加氢性能具有显著影响. 因此, Pd-Cu(111)和Zn-Cu(111)表面加氢能垒较高(表3中反应R3~R5), 加氢活性较低, 可能是由于其表面铜原子的带中心过高或过低导致的[31,32]. 而Ag-Cu(111)和Cu(111)表面Cu原子则具有适宜的带中心, 有利于加氢反应R3~R5, 促进了CH3OOCCH2OH的生成, 与文献[33]报道一致.

Table 6 Charge transfer(Q) between DMO, MG and Cu(111), Ag⁃Cu(111) and Zn⁃Cu(111) catalysts

Fig.8 d⁃Partial density of states of Cu in Cu(111)(A), Ag⁃Cu(111)(B), Zn⁃Cu(111)(C) and Pd⁃Cu(111)(D) catalysts
由表7可见, Cu2O(111), Ag-Cu2O(111), Zn-Cu2O(111)和Pd-Cu2O(111)催化剂表面与DMO分子之间转移电荷量分别为0.0934|e|, 0.0917|e|, 0.0913|e|和0.1507|e|, MG分子带有的电荷量分别为0.0129|e|, 0.0048|e|, -0.0047|e|和-0.0002|e|. 众所周知, 催化剂的活性与价带偏态密度(PDOS)强度和带隙大小息息相关. 通常, 能带带隙越小、 价带强度越高, 催化剂的活性越高[34,35]. 图9为 Cu2O(111)和M-Cu2O(111)(M=Ag, Zn, Pd)催化剂表面第一层原子的带偏态密度图. 计算得到Cu+超胞第一层原子价带强度从高到低依次为 Ag-Cu2O(111)(109.8 eV)>Zn-Cu2O(111)(94.3 eV)>Pd-Cu2O(111)(92.6 eV)>Cu2O(111)(87.7 eV). 从图10可以看出, 催化剂的能带间隙越大, 其DMO加氢反应的速控步骤的能垒越高. 对于4种Cu+催化剂来说, DMO加氢生成MG的活化能与催化剂的能带间隙成正相关关系. 综合价带强度和能带带隙2个方面分析可知, Ag-Cu2O(111)催化剂在DMO加氢反应中优异的催化活性可归因于其高价带强度和低能带间隙.

Table 7 Charge transfer(Q) between DMO, MG and Cu2O(111), Ag-Cu2O(111) and Zn⁃Cu2O(111) catalysts

Fig.9 d⁃Partial density of states of atoms of first layer in Cu2O(111)(A), Ag⁃Cu2O(111)(B), Zn⁃Cu2O(111)(C) and Pd⁃Cu2O(111)(D)
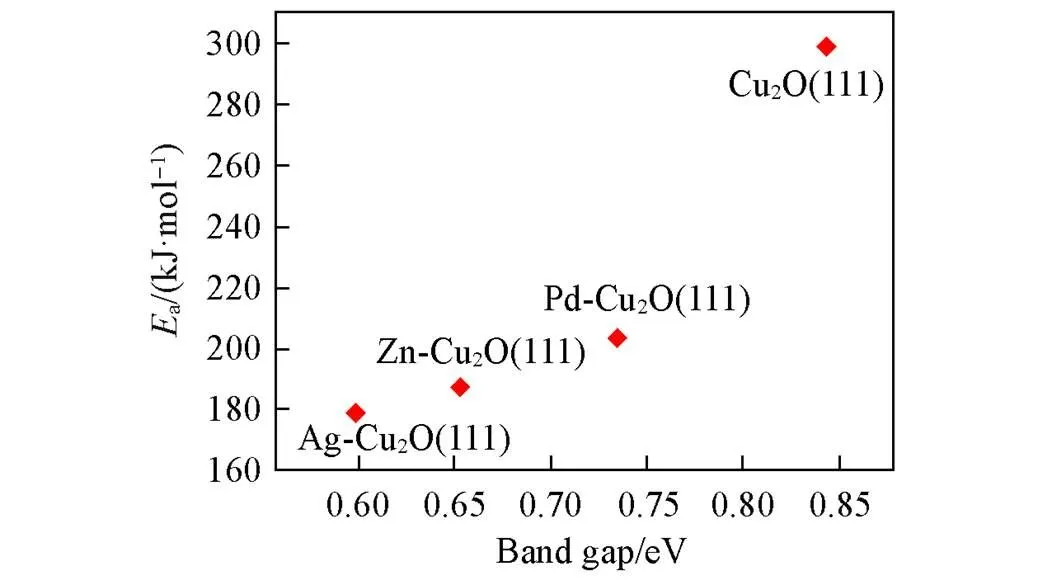
Fig.10 Ea of rate⁃determining step vs. band gap of Cu2O(111) and M⁃Cu2O(111) catalysts
3 结 论
采用DFT计算方法构建了Cu(111)和Cu2O(111)活性晶面, 探讨了掺杂Ag, Zn, Pd原子的Cu(111)和Cu2O(111)晶面上DMO加氢反应机理, 分析了金属掺杂对催化性能的影响. 掺杂Zn助剂可以有效阻止MG的进一步加氢, 提高了MG的选择性, Ag助剂可以有效地提高加氢活性, 而Pd助剂不能有效地控制加氢程度, 降低了MG的选择性. Ag-Cu(111)表面具有适宜的带中心, 生成CH3OOCCH2OH的活性最高. 在Ag, Zn, Pd原子掺杂的Cu2O(111)表面, Ag-Cu2O(111)能带带隙小、 价带强度高, 在DMO加氢反应中具有最佳的催化活性. 提出了Cu催化剂结构调变和催化性能调控的理论方法和基本框架, 为催化剂的设计在理论上提供可靠的指导.
[1] Yang X., Liu B. Y.,, 2014,(9)27—30(杨西, 刘宝勇. 上海化工, 2014,(9), 27—30)
[2] Zhao Y. J., Zhao S., Wang B., Lu J., Ma X. B.,, 2013,(4), 721—731(赵玉军, 赵硕, 王博, 吕静, 马新宾. 化工进展, 2013,(4), 721—731)
[3] Wang B. W., Xu Q., Song H., Xu G. H.,2007, 78—80
[4] Li J. H., Duan X. P., Lin H.Q.,Ye L. M., Yuan Y. Z.,, 2014,(9), 985—994(李建辉, 段新平, 林海强, 叶林敏, 袁友珠. 石油化工, 2014,(9), 985—994)
[5] Tian K. S., Wang B. W., Xu G.H.,., 2006,6), 60—63(田克胜, 王保伟, 许根慧. 天然气化工, 2006,(6), 60—63)
[6] Ye R. P., Lin L., Chen C., Yang J. X., Li F., Zhang X., Li D. J., Qin Y. Y., Zhou Z. F., Yao Y. G.,., 2018,(4)3382—3394
[7] Wang D. H., Zhang C. C., Zhu M. Y., Yu F., Dai B.,, 2017,(17), 4823—4829
[8] An J. W., Wang X. H., Zhao J. X., Jiang S. H., Quan Y. H., Pei Y. L., Wu M. M., Ren J.,, 2019,,110667
[9] Zhao Y. J., Kong L. X., Xu Y. X., Huang H. J., Yao Y. Q., Zhang J. W., Wang S. P.,, 2020,, 12381—12388
[10] Yin A., Wen C., Guo X., Dai W., Fan K.,, 2011,(1), 77—88
[11] Zheng X., Lin H., Zheng J., Duan X., Yuan Y.,, 2013,(12), 2738—2749
[12] Zhang C. C., Wang D. H., Zhu M. Y., Yu F., Dai B.,, 2016,(11), 2857—2863
[13] Wang Q., Qiu L., Ding D., Chen Y. Z., Shi C. W., Cui P., Wang Y.,, 2018,, 68—72
[14] Delley B., Zu C., 1996,(15), 6107—6110
[15] Perdew J. P., Burke K., Ernzerhof M.,, 1996,(18), 3865—3868
[16] Ernzerhof M., Scuseria G. E.,, 1999,, 5029—5036
[17] Dolg M., Wedig U., Stoll H.,1986,, 866—872
[18] Wang Y. Z., Wisesa P.,, 2020,, 110100
[19] Halgren T. A., Lipscomb W. N.,, 1977,(12), 225—232
[20] Hirshfeld F. L.,., 1977,, 129—138
[21] Zhao Y. T., Cui C. N., Han J. Y., Wang H., Zhu X. L., Ge Q. F.,, 2016,(32), 10191—10198
[22] Yue H., Ma X., Gong J.,, 2014,(5), 1483—1492
[23] Li S. M., Wang Y., Zhang J., Wang S. P., Xu Y., Zhao Y. J., Ma X. B.,, 2015,(4), 1243—1250
[24] Hui S. G., Zhang B., Zhang S. H., Li W.,, 2012,(6), 753—758
[25] Zhang R. G., Zhang J., Jiang Z., Wang B. J., Fan M. H.,, 2018,, 732—746
[26] Yang B., Burch R., Hardacre C., Headdock G., Hu P.,, 2012,(6), 1027—1032
[27] Zhang R. G., Zhang J., Zhao B., He L., Wang A. J., Wang B. J.,, 2017,(50), 27936—27949
[28] Larmier K., Liao W. C., Tada S., Lam E., Verel R., Bansode A., Urakawa A., Comas⁃Vives A., Copéret C.,2017,(9), 2318—2323
[29] Li J., Croiset E., Ricardez⁃Sandova L.,, 2012,, 103—114
[30] Huo C. F., Li Y. W., Wang J. G., Jiao H. J.,, 2009,(41), 14713—14721
[31] Zhang R., Wang B., Ling L., Liu H., Huang W.,., 2010,(4), 1175—1180
[32] Kumar A., De Leeuw N. H.,, 2016,, 96—106
[33] Zhang R. G., Zhao B., He L., Wang A. J., Wang B. J.,, 2018,(25), 17487—17496
[34] Zuo Z. J., Ramírez P. J., Senanayake S. D., Liu P., Rodriguez J. A.,2016,(42), 13810—13813
[35] Liu Z. M., Ma L., Junaid A. S. M.,, 2010,(10), 4445—4450
Effects of Ag, Zn, Pd-doping on Catalytic Performance of Copper Catalyst for Selective Hydrogenation of DMO
SONGYouwei, ANJiangwei, WANGZheng, WANGXuhui, QUANYanhong, RENJun, ZHAOJinxian*
(,,030024,)
The active crystal planes of Cu(111) and Cu2O(111) doped with Ag, Zn and Pd atoms were constructed using a density functional theory to investigate the effects of different metal doping on the activity and selectivity for dimethyl oxalate(DMO) hydrogenation to methyl glycolate(MG). The results showed that Zn-doping can prohibit the deep hydrogenation of MG effectively, and Ag additves could effectively improve hydrogenation activity for DMO. Conversely, the addition of Pd increased the energy barrier of MG generation, thus reduced MG selectivity. The Ag-Cu(111) surface has a suitable-center and has the highest activity to generate CH3OOCCH2OH. On the surface of Ag, Zn, Pd atom doped Cu2O(111), Ag-Cu2O(111) has a small band gap and high valence band strength, and has the best catalytic activity in DMO hydrogenation reaction. Based on the above results, theoretical methods for structural modulation and performance regulation of copper-based catalysts are proposed, which provide reliable theoretical guidance for the design of highly efficient catalysts.
Dimethyl oxalate(DMO); Selective hydrogenation; Cu-based catalyst; Metal-doped
O643.3
A
10.7503/cjcu20210842
2021-12-20.
网络首发日期: 2022-04-04.
赵金仙, 女, 博士, 副教授, 主要从事甲醇下游化学品及二氧化碳化学利用方面的研究. E-mail: zhaojinxian@tyut.edu.cn
国家自然科学基金(批准号: 21808154)和山西省应用基础研究项目(批准号: 201901D211059)资助.
the National Natural Science Foundation of China(No. 21808154) and the Key Research and Development Program of Shanxi Province, China(No. 201901D211059).
(Ed.: V, K, S)
