CoFe2O4/g-C3N4对HMX 和TKX-50 的催化分解特性
2022-07-13陈苏杭徐抗震
万 冲,王 晨,陈苏杭,徐抗震
(西北大学化工学院,陕西 西安 710069)
1 引言
固体推进剂作为固体火箭的动力源,其能量的增加和燃烧性能的改善可以显著提升导弹武器装备和固体助推器的机动能力、载荷和射程。改性双基推进剂、复合推进剂和硝酸酯增塑聚醚(NEPE)推进剂因其综合性能良好而被广泛应用于武器弹药中[1-4]。固体推进剂主要由氧化剂、燃料和黏合剂等组成,由于氧化剂所占比例大,且其热分解速率和效率直接影响推进剂的燃烧性能和弹道性能,所以降低氧化剂的分解温度、提高其放热量是固体推进剂的重要研究目标[5-8]。燃烧催化剂作为固体推进剂的另一重要组分[9-10],能够降低氧化剂的分解温度和活化能、提高其放热量,在调控推进剂燃烧压力指数和高压下平台燃烧效果上起着不可或缺的重要作用,因此开发低成本高效固体推进剂燃烧催化剂具有重要意义。目前,燃烧催化剂主要包括金属配合物、二茂铁类、新型碳材料以及金属氧化物等[9],其组成、结构、形貌均直接影响催化活性[11-12]。纳米金属复合氧化物,如尖晶石类CuFe2O4和CoFe2O4及钙钛矿类Bi2WO6和PbZrO3等,其多金属相互掺杂,易引起电荷转移,产生空位、发生晶格畸变,活性位点多,催化性能较好,有望代替金属氧化物等作为固体推进剂的燃烧催化剂[13-16]。
二维石墨相氮化碳(g-C3N4)作为一种新型聚合物半导体材料,具有类似于氧化石墨烯(GO)的二维平面片状结构,可由尿素或三聚氰胺经高温直接煅烧而得。因制备过程简单、成本低、稳定性好、比表面积大、吸收光谱范围广、催化活性高、热稳定性好等优点在半导体、发光材料、催化剂载体和储能材料等领域已广泛应用[17-20],并且在含能材料领域也展现一定的应用前景。Li 等[21]首 次 将g-C3N4作 为 催 化 剂 应 用 于 高 氯 酸铵(AP)的 催 化 分 解。Tan 等[22]通 过 共 沉 淀 法 将g-C3N4与CuO 直接复合制备了CuO/g-C3N4二元材料,发现其对AP 的催化分解能力随CuO 含量的增加而增强。Zhang 等[23]将CuO 与改性后的g-C3N4复合,进一步提高其对AP 的催化能力。本课题组也进行了相关研究,分别制备了CuFe2O4/g-C3N4和Bi2WO6/g-C3N4二元复合材料,考察了其对AP、黑索今(RDX)及哈托(TKX-50)等的催化分解性能并分析了其热分解机理,结果表明g-C3N4作为载体能够增进纳米金属复合氧化物的催化性能,代替GO 在固体推进剂领域展现出了更好的应用前景[24-25]。
综上,为了进一步确定g-C3N4对纳米金属氧化物催化性能的增强作用,本研究采用原位生长制备CoFe2O4/g-C3N4复合材料,研究其结构形貌和对HMX及TKX-50 催化分解的影响,并与CoFe2O4进行比较,为g-C3N4在固体推进剂领域的应用进一步提供参考。
2 实验部分
2.1 试剂与仪器
试剂:六水合硝酸钴[Co(NO3)2·6H2O]、九水合硝酸铁[Fe(NO3)3·9H2O]、无水乙酸钠、聚乙二醇2000、乙二醇、尿素、无水乙醇均为市购分析纯;HMX和TKX-50 由西安近代化学研究所提供。
仪器:X 射线粉末衍射仪(XRD,日本理学);傅里叶红外光谱仪(FT-IR,日本岛津);场发射扫描电镜(SEM,德国卡尔蔡司);Autosorb-iQ 自动气体吸附系统(BET,美国麦克);差示扫描量热仪(DSC,德国耐驰)。
2.2 样品的制备
2.2.1 g-C3N4的制备
g-C3N4采用煅烧法制备[19]:取20 g 尿素在550 ℃下煅烧3 h(连续煅烧2 次),将得到的块状样品研磨并用去离子水洗涤3 次后,置入一定量的去离子水中超声处理10 h,离心过滤并于60 ℃下真空干燥,从而制得所需g-C3N4样品。
2.2.2 CoFe2O4的制备
CoFe2O4采 用 溶 剂 热 法 制 备:将Fe(NO3)3·9H2O(1.33 g)溶于60 mL乙二醇中,缓慢加入Co(NO3)2·6H2O(1.46 g),超声处理30 min后,加入无水乙酸钠(7.2 g)和聚乙二醇(2 g)并搅拌12 h,将得到的均相溶液转移至100 mL 聚四氟乙烯内衬的不锈钢高压反应釜中,200 ℃下反应18 h。待反应釜自然冷却至室温后,将产物经无水乙醇和去离子水离心清洗,60 ℃下干燥12 h 得到CoFe2O4纳米颗粒。
2.2.3 CoFe2O4/g-C3N4的制备
相比于制备CoFe2O4,先将g-C3N4(50 mg)分散于乙二醇溶液(60 mL)并超声3 h,然后再加入Fe(NO3)3·9H2O(1.33 g)和Co(NO3)2·6H2O(1.46 g),后续步骤与制备CoFe2O4相同,最终得到CoFe2O4/g-C3N4样品。
3 结果与讨论
3.1 表征分析
采 用XRD 和FT-IR 对 所 制 备 的CoFe2O4和CoFe2O4/g-C3N4进行物相分析,结果如图1 所示。由图1a 可知,在18.29°、30.08°、35.44°、43.06°、47.15°、53.45°、56.79°和62.59°处的衍射峰分别对应于CoFe2O4标准卡片JCPD#22-1086 的(1 1 1)、(2 2 0)、(3 1 1)、(4 0 0)、(3 3 1)、(5 1 1)和(4 4 0)晶面[27]。CoFe2O4/g-C3N4与CoFe2O4的XRD 衍射峰位置几乎完全一致,表明引入g-C3N4没有改变CoFe2O4的晶型。但由于g-C3N4的含量较少,其衍射峰相对于CoFe2O4的衍射峰十分微弱,因此在CoFe2O4/g-C3N4中未能观察到g-C3N4的衍射峰。由图1b 可知,在CoFe2O4/g-C3N4中除了有CoFe2O4的吸收峰之外,在1250~1650 cm-1范围内的吸收峰对应于C—N 和C=N 杂环固有的伸缩振动吸收,在802 cm-1处的强峰对应于三嗪环的振动吸收[24],与g-C3N4的振动吸收一致。
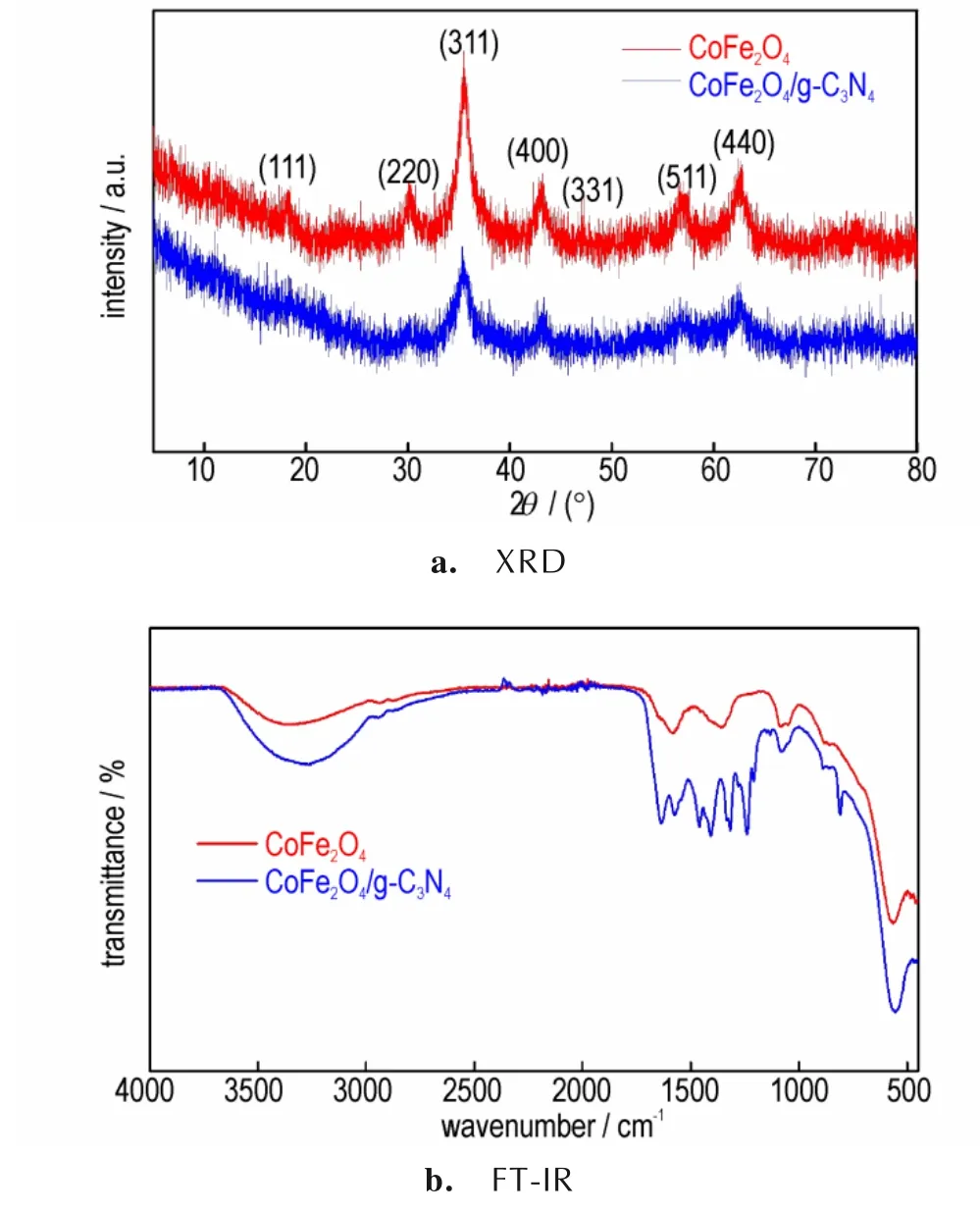
图1 CoFe2O4和CoFe2O4/g-C3N4的XRD 和FT-IR 结果Fig.1 XRD and FT-IR patterns of CoFe2O4and CoFe2O4/g-C3N4
为了进一步确定CoFe2O4颗粒是否原位生长到g-C3N4载体上,采用SEM 分别观察了g-C3N4,CoFe2O4和CoFe2O4/g-C3N4的微观形貌,并对CoFe2O4/g-C3N4进行了表面元素分布分析,结果如图2 所示。由图2a~2b 可 见,原 始 的g-C3N4为 片 层 状 结 构,而CoFe2O4颗粒结构致密,但存在明显的团聚现象。图2c 显示了CoFe2O4均匀地负载在g-C3N4的表面,界面之间连接十分紧密。作为载体,g-C3N4有效解决了CoFe2O4的团聚问题,使得纳米颗粒分布更加均匀。CoFe2O4/g-C3N4的EDS 结果和元素分布图(图2d~2i)表明:C、N、O、Fe、Co 等元素均匀分布在复合材料的表面,进一步说明了CoFe2O4/g-C3N4复合材料的成功制备。

图2 g-C3N4,CoFe2O4和CoFe2O4/g-C3N4的SEM 图(a~c);CoFe2O4/g-C3N4的元素分析结果和元素分布图(d~i)Fig.2 SEM images of g-C3N4,CoFe2O4 and CoFe2O4/g-C3N4(a-c);element analysis results and element distribution diagram of CoFe2O4/g-C3N4(d-j)
比表面积是评估催化剂活性的一个重要指标[20],对CoFe2O4和CoFe2O4/g-C3N4进行氮吸附比表面积测定,结果如图3 所示。由图3 可知,CoFe2O4的吸附-脱附曲线在p/p0为0.8~1.0 范围内有1 个很小的H3型滞回 环,而CoFe2O4/g-C3N4在p/p0为0.4~1.0 范 围 内 有1 个非常明显的H3型滞回环,表明CoFe2O4/g-C3N4存在更多的介孔。同时,CoFe2O4/g-C3N4的比表面积(229.83 m2·g-1)显 著 大 于CoFe2O4的 比 表 面 积(196.38 m2·g-1),CoFe2O4/g-C3N4的孔径最大可达~180 nm,显著大于CoFe2O4的孔径(~100 nm),充分表明g-C3N4载体的引入,使材料的孔含量增多,比表面积和孔径增大,从而有利于提高催化活性。

图3 CoFe2O4 和CoFe2O4/g-C3N4 的氮气吸附-脱附曲线、孔径分布曲线和比表面积Fig.3 Nitrogen adsorption-desorption curves,aperture distribution curves and specific surface area of CoFe2O4 and CoFe2O4/g-C3N4
3.2 对HMX 的催化分解活性研究
起始分解温度、峰温是评估催化分解性能的重要指标参数[15]。为了研究CoFe2O4和CoFe2O4/g-C3N4对HMX 的催化活性,在相同条件下采用DSC 分别测试了二者对HMX 的热分解行为影响。将催化剂样品分别与HMX 以1∶4 的质量比进行混合,氮气气氛下,流速40 mL·min-1,测试样品用量大约为0.5 mg,升温速率为10 ℃·min-1,结果如图4 所示。HMX 的放热分解是一个熔融分解过程,峰温为283.2 ℃。 加入CoFe2O4和CoFe2O4/g-C3N4后,HMX 的分解峰形发生显著改变,吸热熔融过程消失,分解温度显著提前,变成一个大而宽的缓慢放热分解峰。CoFe2O4+HMX 和CoFe2O4/g-C3N4+HMX 起始分解温度由纯HMX 的281.3 ℃提前至257.2 ℃和254.9 ℃,峰温由283.2 ℃提 前 至277.6 ℃和276.2 ℃。 与CoFe2O4相 比,CoFe2O4/g-C3N4使HMX 分解的峰温提前了1.4 ℃,表现出更好的性能。其原因是引入g-C3N4后有效地改善了CoFe2O4的团聚,并增大了CoFe2O4的比表面积。
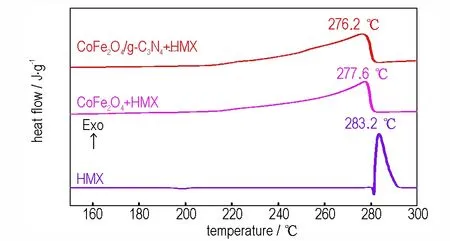
图4 HMX,CoFe2O4+HMX 和CoFe2O4/g-C3N4+HMX 的DSC曲线Fig.4 DSC curves of HMX,CoFe2O4+HMX and CoFe2O4/g-C3N4+HMX
含能材料的表观活化能是体现分解过程难易的一个重要指标,对研究催化分解性能具有重要意义[24]。为了对比CoFe2O4和CoFe2O4/g-C3N4对HMX 的催化分解的影响,分别对CoFe2O4+HMX 和CoFe2O4/g-C3N4+HMX在不同升温速率下(5、10、15 ℃·min-1和20 ℃·min-1)进行DSC 测试,结果如图5 所示。由图5 可知,随着升温速率增加,分解峰的峰形基本不变,但依次向高温推移。利用Kissinger法计算了热分解过程的动力学参数,结果见表1。由表1 可知,与HMX 的表观活化能相比(502.2 kJ·mol-1)[27],CoFe2O4+HMX 和CoFe2O4/g-C3N4+HMX 的 表 观 活 化 能 分 别 降 低 了276.6 kJ·mol-1和341.1 kJ·mol-1,活化能显著降低,表明2 种催化剂均表现出良好的催化分解作用。同时,CoFe2O4/g-C3N4+HMX 的表观活化能显著低于CoFe2O4+HMX 的表观活化能(相差64.5 kJ·mol-1),表明CoFe2O4/g-C3N4比CoFe2O4呈现更好的催化分解作用。因此,g-C3N4的引入进一步提高了CoFe2O4的催化性能。
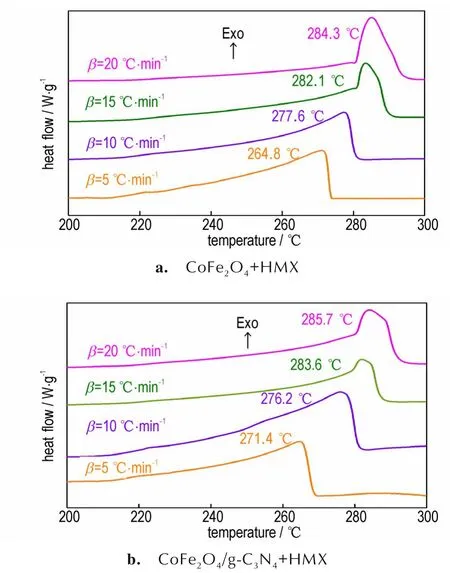
图5 CoFe2O4+HMX 和CoFe2O4/g-C3N4+HMX 在不同升温速率下的DSC 曲线图Fig.5 DSC curves of CoFe2O4+HMX and CoFe2O4/g-C3N4+HMX at different heating rates
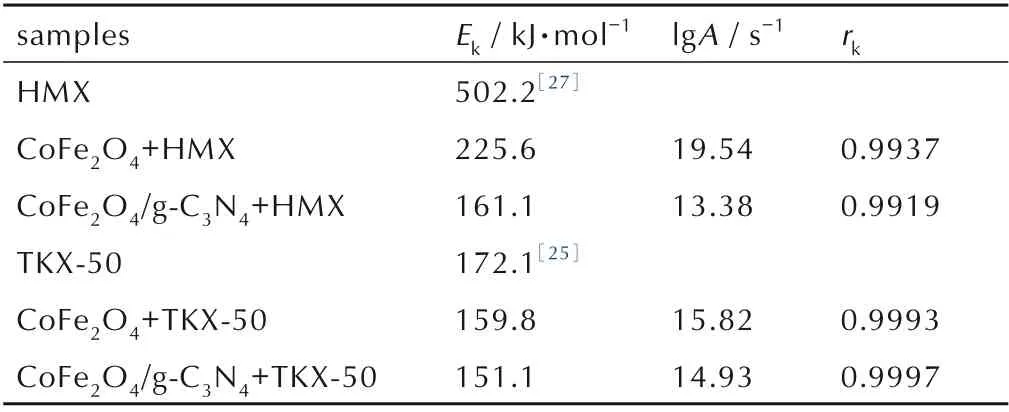
表1 样品的分解动力学参数Table.1 Kinetic parameters of decomposition for samples.
3.3 对TKX-50 的催化分解活性研究
基于CoFe2O4/g-C3N4对HMX 良好的催化分解作用,采用相同的方法研究了其对TKX-50 的热分解影响,结 果 如 图6 所 示。 由 图6 可 知,CoFe2O4和CoFe2O4/g-C3N4均能使TKX-50 的热分解温度显著降低,使TKX-50 的峰温从238.2 ℃降低至199.3 ℃和196.9 ℃,分别降低了38.9 ℃和41.3 ℃。同时,TKX-50 加入催化剂前后的峰型基本没有发生变化,均为1 个剧烈的放热分解过程和随后紧接的1 个缓慢放热分解过程,只是加入催化剂后放热峰的位置大幅前移,表明催化剂的加入并没有改变TKX-50的分解过程,只是使其分解更加容易。CoFe2O4/g-C3N4+TKX-50 的峰温比CoFe2O4+TKX-50 的峰温低了2.4 ℃,也进一步证实CoFe2O4/g-C3N4比CoFe2O4对TKX-50 有更好的催化分解作用。

图6 TKX-50,CoFe2O4+TKX-50 和CoFe2O4/g-C3N4+TKX-50的DSC 曲线Fig.6 DSC curves of TKX-50,CoFe2O4+TKX-50 and CoFe2O4/g-C3N4+TKX-50
CoFe2O4+TKX-50 和CoFe2O4/g-C3N4+TKX-50 在不同升温速率(5、10、15 ℃·min-1和20 ℃·min-1)下的DSC 结果如图7 所示。由图7 可见,分解峰随升温速率增加的依次向高温推移,峰形保持不变。同样通过 Kissinger 法 计 算 得 到 了 CoFe2O4+TKX-50 和CoFe2O4/g-C3N4+TKX-50 的热分解动力学参数,结果见表1。由表1可知,加入催化剂CoFe2O4和CoFe2O4/g-C3N4后,TKX-50的分解表观活化能分别降低至159.8 kJ·mol-1和151.1 kJ·mol-1,相 比 于TKX-50 的 表 观 活 化 能(172.1 kJ·mol-1)[25],分 别 降 低 了12.3 kJ·mol-1和21.0 kJ·mol-1,均表现出良好的催化活性。同时,与CoFe2O4相比,引入g-C3N4后表观分解活化能进一步降 低 了8.7 kJ·mol-1,证 明CoFe2O4/g-C3N4呈 现 出 比CoFe2O4更好的对TKX-50 催化性能。
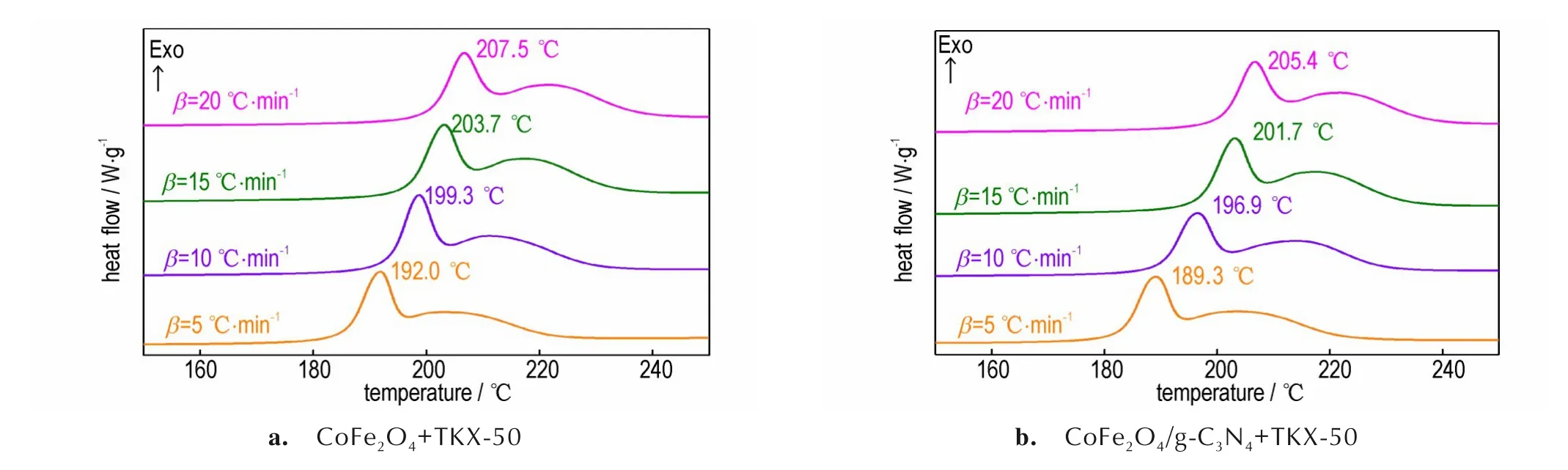
图7 CoFe2O4+TKX-50 和CoFe2O4/g-C3N4+TKX-50 在不同升温速率下的DSC 曲线Fig.7 DSC curves of CoFe2O4+TKX-50 and CoFe2O4/g-C3N4+TKX-50 at different heating rates
3.4 残渣分析
利用SEM 对比分析了CoFe2O4/g-C3N4+HMX 和CoFe2O4/g-C3N4+TKX-50 分解前后的形貌变化,并对CoFe2O4/g-C3N4+TKX-50 热分解后的残渣进行了表面元素分布分析结果如图8 所示。从图8a 和图8d 中可以 看 出,HMX 和TKX-50 与CoFe2O4/g-C3N4均 匀 复 合在一起。受热分解后,CoFe2O4/g-C3N4+HMX 形成了纳米级大小不均匀的块状物(图8b),进一步放大发现(图8c),由于HMX 在催化剂CoFe2O4/g-C3N4的作用下分解比较彻底,残渣只观察到CoFe2O4/g-C3N4的存在。而对于TKX-50,在测试温度范围内,其分解不够彻底,形成了分解残渣和CoFe2O4/g-C3N4的微米级块状混合物(图8e)。由图8f 可知,EDS 元素分析结果发现有很高的C、N 元素存在,因此TKX-50 在低温下的催化分解不是很完全。

图8 CoFe2O4/g-C3N4+HMX、CoFe2O4/g-C3N4+TKX-50 混合后的SEM(a,d);热分解后残渣的SEM 图(b,c,e);CoFe2O4/g-C3N4+TKX-50 热分解后残渣的EDS 结果(f)Fig.8 SEM images of CoFe2O4/g-C3N4+HMX(a)and CoFe2O4/g-C3N4+TKX-50(d);the SEM imagesof residue after thermal decomposition of mixed materials(b,c,e);the EDS results of CoFe2O4/g-C3N4+TKX-50 residue after thermal decomposition(f)
4 结论
(1)采用溶剂热法成功制备了CoFe2O4/g-C3N4二元复合材料,CoFe2O4纳米颗粒均匀地负载在g-C3N4表面,二者结合紧密。引入g-C3N4没有改变CoFe2O4的晶型,改善了CoFe2O4纳米颗粒的团聚问题,显著增加了比表面积;
(2)CoFe2O4/g-C3N4呈现出了优异的催化分解性能,CoFe2O4/g-C3N4使HMX 和TKX-50 的热分解峰温分别降低了7.0 ℃和41.3 ℃,表观活化能分别降低了341.1 kJ·mol-1和21.0 kJ·mol-1;
(3)相比于CoFe2O4,g-C3N4载体的引入使得HMX和TKX-50的热分解峰值温度分别降低了1.4 ℃和2.4 ℃,表观活化能分别降低了64.5 kJ·mol-1和8.7 kJ·mol-1,因此CoFe2O4/g-C3N4比CoFe2O4呈现更优的催化分解性能;
(4)HMX 在催化剂作用下受热分解比较彻底,而TKX-50 受热分解后不够完全,并与CoFe2O4/g-C3N4形成了微米级的块状混合物。
