牦牛测序数据缺失值填充的方法比较
2022-07-07王嘉博益西康珠钟金城
秦 婕,王嘉博,益西康珠,钟金城
(1.青藏高原动物遗传资源保护与利用教育部重点实验室,四川成都 610041;2.青藏高原动物遗传资源保护与利用四川省重点实验室,四川成都 610041)
全基因组测序(Whole Genome Sequencing,WGS)是对细胞或生物体所有的基因组进行测序,获得完整的基因组信息.全基因组测序数据可以找到基因与表型之间的联系,在挖掘动植物重要经济性状相关的功能基因、分析遗传机制等方面有重要意义.
我国牦牛主要分布在青藏高原海拔2 500米以上的高寒草原及高山峡谷区域,有着耐高寒和耐低氧的优良性状,是牧民生活依赖的重要生产资料[1-3].由于传统牦牛饲养方式造成的长期过度繁殖和近亲繁殖,家养牦牛的繁殖能力、生长速度、成年体型和产奶量下降,给当地畜牧业的发展造成了影响[4]. 对全基因组测序数据进行分析,可以找到与生长性状相关的候选基因,但是牦牛的全基因组测序数据一般有较多缺失值,需要利用基因型填充的方法填充缺失值.
基因型填充方法的基本原理是根据参考群体提供的基因型信息,构建出参考群体和目标群体之间共享的单倍型信息,比对目标群体与参考群体之间共享的单倍型信息,将目标群体缺失的基因型信息填充完整,得到完整的基因型数据[5]. 基因型填充方法大致分为两类,一类是计算密集型的,如IMPUTE、MACH和fastPHASE 等,还有一类是计算高效型,如PLINK、MINIMAC 和BEAGLE 等. 计算密集型的基因型填充软件在填充的过程中充分考虑到所有已知的基因型信息,使得对未知的基因型信息的估算更加精确,但是填充耗时较长.而计算高效型的基因型填充软件在填充过程中仅仅关注与特定的SNP 位点相邻的一部分标记的基因型信息,填充耗时较短但是填充准确性相对要低一点. 在日本黑牛的研究中,利用BEAGLE软件从缺失基因型的数量、参考群体的大小、参考群体和目标群体之间的遗传关系评估了日本黑牛测序数据的填充准确性.对于不同数量的缺失基因型的影响,50 K、26 K 和20 K 的测序数据的相关性都比较高,但7 K 的相关性较低;对于参考群体的大小的影响,50 K、26 K 和20 K 的测序数据在参考群体超过400 时的相关性趋于平稳,但当参考群体数量增加到400 以上时,7 K 测序数据的相关性略有增加;对于参考群体和试验群体之间遗传关系的影响,使用相关参考群体的相关性高于使用无关参考群体的相关性[6].在肉牛的研究中,使用 BEAGLE、FIMPUTE 和 IMPUTE2 软件对多品种的肉牛群体进行了填充. FIMPUTE 软件对纯种群体填充的填充匹配率在94.20%到 97.93% 之 间, IMPUTE2 软 件 为 95.35% 到98.31%,BEAGLE 软件为90.02%到96.38%.杂交动物的填充匹配率为54.15% ~97.53% (FIMPUTE),57.04% ~ 97.46% (IMPUTE2), 以 及 54.35% ~95.64%(BEAGLE)[7].在牛的研究中,选择三种参考基因组,对数据进行基因分型后,利用PEDIMPUTE、FINCHAP、FIMPUTE 和 BEAGLE 软件进行填充,FIMPUTE 软件的填充匹配率最高,约为95%,其次是BEAGLE 软件,约为92%,另外两种软件的填充匹配率受基因分型的影响变化较大[8].基因型填充技术在人类[9]、鸡[10]、羊[11]、猪[12]、植物[13]上都有广泛的应用.
本文将利用StochasticImpute 函数、impute. knn 算法和BEAGLE 软件三种基因型填充方法对牦牛的测序数据进行填充,探究三种填充方法在不同的缺失率条件下的填充效果,以填充匹配率、相关性和填充耗时为评价指标,期望得到填充效果更好的填充方法,用于后续的研究中.
1 材料与方法
1.1 数据处理
本文所用的牦牛基因型数据来源于已发表的文章(JIA C,Animal Genetics,2020)[14],文章测量了青海大通牧场354 头雌性阿什旦牦牛的4 个生长性状,并对所有个体的全基因组测序数据进行了基因分型和质量控制,得到了三个类型的文件,分别是ped 文件、map 文件和 csv 文件.其中 ped 文件包含了 354 头牦牛的98 688 个SNP 位点的信息,map 文件包含了354 头牦牛的遗传图谱信息,csv 文件包含了354 头牦牛体重、肩高、体长和胸围4 个生长性状的信息.本研究将ped 文件转换成hapmap 格式基因组信息文件.首先,用 PLINK 软件将 ped 文件转换为 0、1、2 型的文件,其中 0、1、2 型分别对应主要(major)等位基因、杂合基因和次要等位基因(minor). 因为本文是对不同的基因型填充方法进行比较,所以需要先用GATK 软件对原始的98 k 数据进行过滤. 从过滤后的98 k 数据中随机选取10 k 数据,总共生成30 组不同的10 k数据作为本文的真实数据集. 将每组数据按照5%、10%、15%和20%的比例进行人为的缺失,缺失的基因型用“NA”表示,把含有缺失基因型的数据作为参考数据集.缺失掉的基因型数据保留在另一份独立文件中作为预测数据集的真实基因型.利用三种基因型填充方法对参考数据集中的缺失基因型进行预测,得到预测数据集.最后比较真实基因型数据和预测基因型数据的匹配率和相关性.
1.2 基因型填充方法
StochasticImpute 函数的填充原理是利用等位基因的频率进行缺失值的填充,即以所有个体为参考群体,计算每个SNP 位点中所有等位基因的频率,用频率最高的等位基因填充该SNP 位点中的缺失值.本研究根据数值型基因型文件,利用StochasticImpute 函数分别计算0、1、2 三种基因型频率,用频率最高的等位基因型作为填充的基因型.
impute. knn 算法需要调用 R 语言中的“impute”软件包,这种算法利用基因型数据中特定数目近邻基因型值来填充含有缺失值的个体的基因型[15]. 首先需要将候选邻居进行分类,再使用距离公式计算含有缺失值的基因与候选邻居之间的距离,其中用来计算距离的基因坐标应为基因中未缺失的元素.对于候选邻居可能缺少用于计算距离的坐标的情况,需要计算非缺失元素的平均值. 找到一个基因的k 个近邻后,根据这k 个近邻中的大部分邻居所属的类别决定含有缺失值的基因型,然后通过对其相邻非缺失元素求平均值来估算缺失元素[16].本文设置的k 值为3,选择了欧几里德计算邻近数据之间的距离.
本文选择BEAGLE 5.1[17]作为填充软件,该软件利用隐马尔可夫模型(Hidden Markov Models,HMM)进行基因型填充.首先利用HMM 计算参考面板的单倍型中一个标记到下一个标记的概率,其中每个标记指的是一段有相同等位基因的单倍型集合[18]. 在每个标记处,用等位基因标记的概率之和作为该等位基因的估算概率.从第一个标记到最后一个标记的概率之和就是特定的单倍型概率. 然后,对目标样本进行基因分型,得到单倍型集合. 再根据目标样本与参考面板之间共有的基因序列建立模型,利用计算得到的参考面板中的等位基因标记的概率,预测目标样本中同样标记处的缺失值,进行填充[17].本文需要将0、1、2 型文件进行转换,将“0”转换为“AA”,“1”转换为“AB”,“2”转换为“BB”,“NA”转换为“??”,得到新的基因型文件,再利用BEAGLE 软件进行填充.
1.3 评价基因型填充方法的标准
为了比较三种基因型填充方法的填充效果,本文将把填充匹配率、相关性和填充耗时作为评价标准.其中填充匹配率指的是填充正确的基因型个数与需要填充的基因型个数的比值. 其中Nmatch是填充后准确预测基因型的数目,Ntotal是所有缺失基因型的数目.

相关性计算为真实的基因型与填充得到的数值型基因型之间的相关性. 其中Gimpute是填充后的基因型,Greal是真实的基因型.

填充耗时指的是从填充开始一直到填充结束所用的时间.用R 语言中的system. time 函数来标记开始和结束时间,最后将软件运行返回的时间作为填充耗时.
2 结果与分析
2.1 填充匹配率的比较
对于基因型数据,随机设置5%、10%、15% 和20%的缺失率,用三种基因型填充方法进行填充,重复30 次,得到填充匹配率,如下图1.
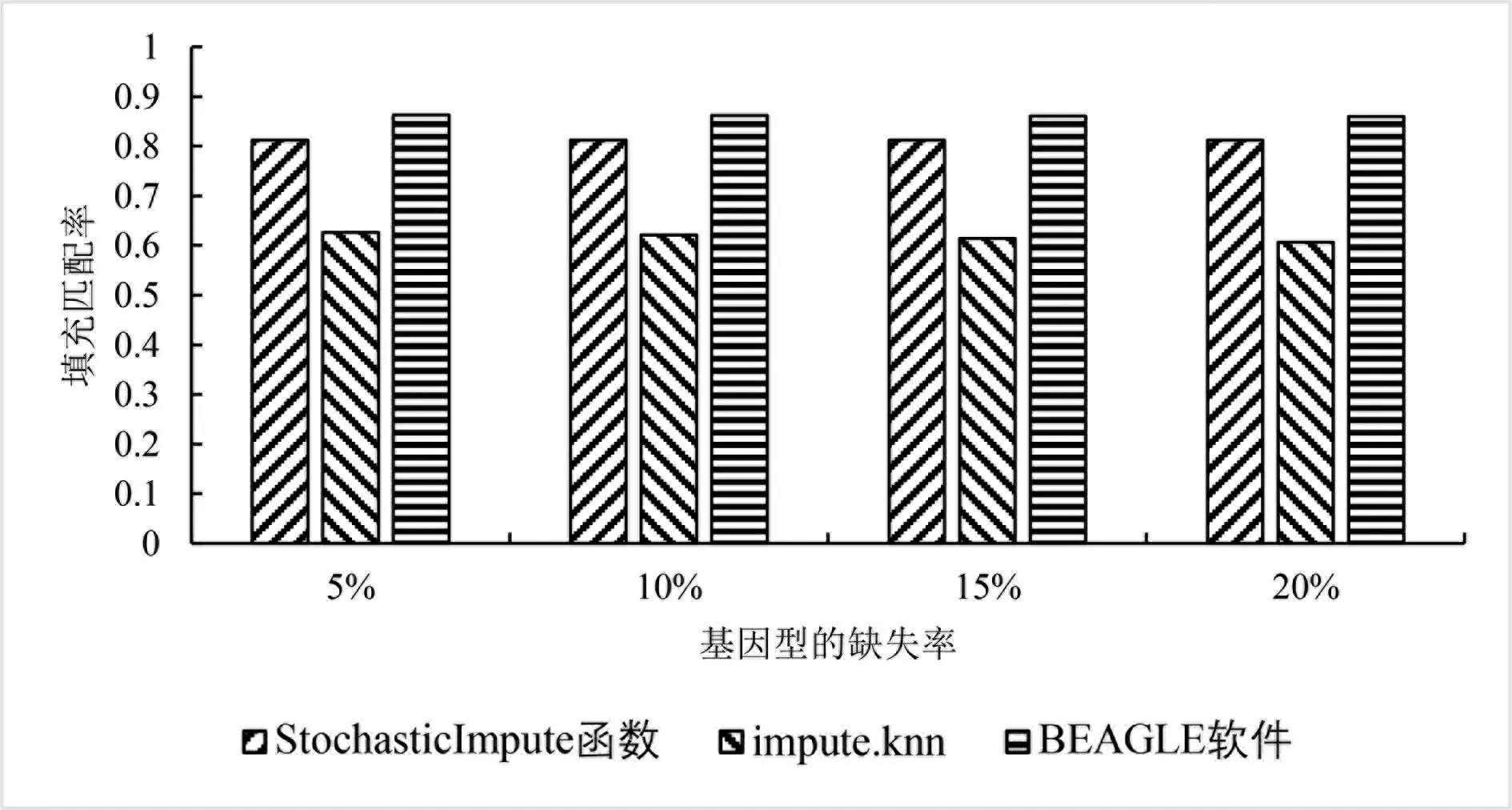
图1 三种填充方法在不同缺失率条件下的匹配率Fig.1 Matching rate of three imputation methods under different missing rate
由图1 可以看出,在不同的缺失率条件下,BEAGLE 软件的填充匹配率最高,填充匹配率分别为0.863 0、0.861 7、0.860 9 和 0.859 9,其次是 StochasticImpute 函数,填充匹配率分别为0.812 5、0.812 3、0.812 5 和0.812 5,填充匹配率最低的是impute. knn算法,填充匹配率分别为0.626 5、0.621 4、0.614 8 和0.606 3. 三种方法的匹配率并不随着缺失率的增大而产生的显著的变化.
2.2 相关性的比较
利用30 次的重复抽样数据,计算真实基因型数据数值与预测基因型数值之间的相关性,以30 次结果的平均值作为考量(图2).
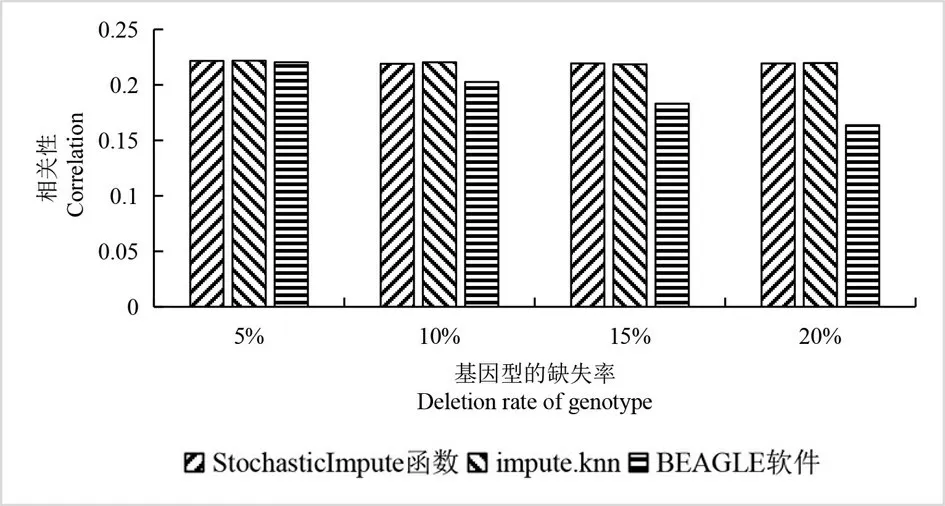
图2 三种填充方法在不同缺失率条件下的相关性Fig.2 Correlation of three imputation methods under different missing rate conditions
由图2 可以看出,在不同的缺失率条件下,StochasticImpute 函数和impute. knn 算法的相关性相对较高,StochasticImpute 函数的相关性分别为0.221 6、0.219 0、0.219 3、0.219 3,而 impute. knn 算法的相关性分别为 0.221 8、0.220 4、0.218 5、0.219 7. BEAGLE 软件的相关性分别为 0.220 4、0.202 6、0.183 1、0.163 7,随着缺失率的增大,BEAGLE 软件的相关性在逐渐降低,并且几乎都低于另外两种填充方法的相关性. 而随着缺失率的增大,StochasticImpute 函数和impute. knn 算法的相关性基本没有变化.
2.3 填充耗时的比较
在利用三种方法进行填充的时候,记录三种填充方法重复30 次得到的填充耗时,取平均值,用对数函数进行标准化,得到图3.
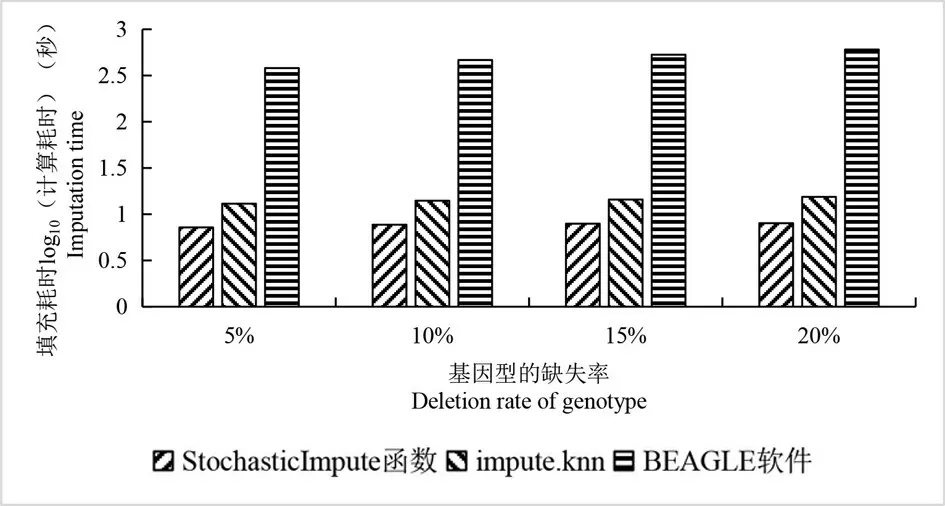
图3 三种填充方法在不同缺失率条件下的填充耗时Fig.3 Impute time of three imputation methods under different deletion rates
由图3 可以看出,BEAGLE 软件的填充耗时最长,填充耗时分别为 380.7 s、465.3 s、531.0 s 和604.5 s,其次是 impute. knn 算法,填充耗时分别为13.0 s、14.0 s、14.4 s 和 15.4 s,填充耗时最少的是StochasticImpute 函数,填充耗时为7.2 s、7.7 s、7.9 s和8.0 s.并且随着缺失率的增大,三种填充方法的填充耗时都在增加,其中BEAGLE 软件的填充耗时增加得最多,在缺失率为20% 的条件下,填充耗时为604.5 s.
3 讨论
因为相关性只考虑数值(即数值型基因型值)之间的顺序关系,在基因型数据用0、1、2 的数值表示时会造成了巨大的偏差.在Korku 等[19]的研究报道中,与本文同样使用的是0、1、2 型的基因型文件,但是填充得到的相关性和填充匹配率都比较高,而且填充匹配率比较高的群体,对应的相关性也比较高. 本文的填充匹配率比较高,但是相关性却比较低,可能是因为数据中的0、1 型的基因型比较多.虽然BEAGLE 软件填充正确的基因型比较多,但是这些基因型大多是0、1 型,也可能对相关性造成影响,导致相关性比较低.
因为StochasticImpute 函数的原理是根据等位基因的频率填充缺失的基因型,每个缺失基因型填充的时候只利用所有参考群体该位置的基因型,所以填充耗时比较短.对于参考群体数量比较多,突变位点比较少的数据,用StochasticImpute 函数的填充效果比较好,填充准确率比较高,填充耗时也比较短.但是StochasticImpute 函数没有考虑到群体内部具体的遗传关系和分层结构,而群体内部具体的遗传关系和分层结构在牦牛数据中尤其重要,因此StochasticImpute 函数不适用于填充牦牛数据.
impute. knn 算法的填充效果与k 值有关,选择合适的k 值,得到的填充效果比较好. 在Di 等[20]的研究中,因为选择了合适的k 值,所以利用impute. knn算法填充得到的填充效果比较好.因为impute. knn 算法的k 值不能通过计算确定,需要人为设置,对于样本量比较小的数据,可以计算所有的k 值找到最合适的值,而本文的样本量比较大,没有办法计算所有的k 值,选择k 值为3,相对于354 个个体而言很少,所以填充效果比较差.impute. knn 算法适合样本量比较小的数据.
在这三种填充方法中,BEAGLE 软件的填充效果比较好,填充匹配率最高,填充耗时也较短. 在Yang等[21]的报道中,利用BEAGLE 软件对水牛的测序数据进行填充,得到的填充匹配率为83%左右. 而本文利用BEAGLE 软件对牦牛的测序数据进行填充得到的填充匹配率为86%左右,填充匹配率更高,说明可以利用BEAGLE 软件对牦牛的基因型数据进行填充.在邓天宇等[22]的研究中,利用BEAGLE 软件填充模拟数据得到的填充耗时和本文的结果比较接近,都相对较短.并且随着缺失率的增加,BEAGLE 软件的填充耗时也在增加,但是增长的趋势较慢.
本文研究了三种不同的基因型填充方法对牦牛的测序数据进行填充的效果,但是只对比了在不同缺失率条件下的结果,对于参考群体大小、目标群体位点比例、SNP 位点数目等方面并未进行研究,希望能够在之后的研究中针对不同的方面展开更多的研究.
因为本文使用的测序数据是由354 头牦牛,10 k测序数据组成,所以在基因型数据相对较小的时候,用BEAGLE 软件进行填充的匹配率比较高,填充耗时也比较少.当需要填充的基因型数据比较多的时候,用BEAGLE 软件进行填充时,填充匹配率会提高,但是填充耗时也会增长.
牦牛群体一般处于半野生的放牧环境下,因此家系和亚群的背景在遗传关系中具有非常强的结构关系.利用这些信息来进一步提升牦牛基因型数据填充的准确性,可能会是一个理想的研究方向.
4 结论
通过对三种填充方法的填充效果进行对比,BEAGLE 软件的填充匹配率更高,impute. knn 算法的相关性更高,StochasticImpute 函数的填充耗时更短.在缺失率小于20%时,基因型填充方法的填充效果较好,建议使用20%的缺失率对测序数据进行过滤,过滤后的数据利用基因型填充技术填充可以得到可信度较高的基因型数据.
