基于G-四链体的PCR-RCA双重扩增技术检测沙门氏菌
2022-07-07刘健慧张先舟张蕴哲李兰茹王红静耿凤珍檀建新
刘健慧,张先舟,张蕴哲,李兰茹,王红静,高 洁,耿凤珍,檀建新,*
(1.河北农业大学食品科技学院,河北 保定 071000;2.河北旅游职业学院,河北 承德 067000;3.河北大学附属医院,河北 保定 071000)
沙门氏菌()是一种革兰氏阴性食源性病原菌,属肠杆菌科,常以家畜、家禽作为传染源。进食被沙门氏菌污染而未煮熟的食品或未消毒的奶制品可诱发呕吐、腹泻等症状。在我国,75%的细菌性食物中毒事件由沙门氏菌造成,因此为保障食品安全,急需快速灵敏的检测手段对其监控。
目前,食源性病原菌的检测手段主要包括传统的微生物学检测、生理生化鉴定和现代的免疫学方法、分子生物学方法等。纯培养方法虽作为检测的金标准,但需要在各种选择性培养基中进行分离培养、形态检测、生化鉴定,检测耗时长,失去了快检的应用意义;免疫学方法虽不需要复杂的仪器,检测准确性好,但制备高特异性单克隆抗体相对困难、成本高,限制了其使用前景;分子生物学方法主要以核酸检测方法为主,常分为聚合酶链式反应(polymerase chain reaction,PCR)、多重PCR、实时PCR等非等温扩增技术和环介导等温扩增、滚环扩增(rolling circle amplification,RCA)、链置换扩增等等温扩增技术。核酸检测技术虽存在较多弊端,如过于敏感导致假阳性和引物设计相对复杂、需要大量筛选验证等,但其检测周期短、特异性强、可进行多种病原菌同时检测,精准定量,故取得了广阔的发展。
核酸信号辅助放大是快检有效、直接的策略。生物素-亲和素连接、酪酰胺信号放大、纳米颗粒信号放大、核酸染料信号放大等技术均已实现广泛应用,但需额外在体系中添加或标记信号放大因子,使实验设计复杂,且灵敏度易受到添加量的干扰。而G-四链体作为富G的单链功能核苷酸,将其自身或互补碱基设计到相关序列中,可开发出多种成本低、有效性强的生物传感器。如水溶性苯并噻唑染料硫黄素T (thioflavin T,ThT)可特异性识别并嵌入G-四链体凹槽中,导致荧光信号显著增强,进而通过检测荧光强度实现信号放大。
本研究先针对沙门氏菌基因设计PCR扩增引物,并在下游引物的5’端标记15个A碱基(15A),通过PCR扩增积累诱发RCA反应的环外“引物”。同时将G-四链体互补序列和15个A碱基设计成含有黏性末端的发卡结构,并连接成哑铃环状DNA,作为RCA扩增的模板。以PCR产物为“桥梁”,将PCR与RCA偶联,当沙门氏菌基因组DNA存在时,启动RCA扩增,不断积累富G序列,最终通过测量双重扩增产物与ThT混合溶液的荧光强度,实现对沙门氏菌的定量检测。该方法在无靶标的扩增体系中无G-四链体生成,能够极大降低背景值,为食品中致病菌的快检提供新策略。
1 材料与方法
1.1 材料与试剂
志贺氏菌(ATCC 12022)、单核细胞性李斯特菌(CICC 21633)、金黄色葡萄球菌(CICC 21600)、铜绿假单胞菌(ABCC 0927)、大肠埃希氏菌(CMCC 44752)、甲型副伤寒沙门氏菌(CICC 21501)、鼠伤寒沙门氏菌(CICC 21484)、丙型副伤寒沙门氏菌(CICC 21512)均由河北农业大学生物工程实验室提供;核酸序列由金唯智生物科技有限公司合成;细菌基因组DNA提取试剂盒、DNA纯化回收试剂盒 天根生化科技有限公司;2×EsMaster Mix(Dye)、PAGE凝胶制备试剂盒 北京康为世纪生物科技有限公司;100 bp DNA ladder、T4 DNA聚合酶 宝生物工程(大连)有限公司;50 bp ladder 北京聚合美生物科技有限公司;2.0 DNA聚合酶 NEB(北京)有限公司;ThT 北京Solarbio公司;其他主要试剂均为国产分析纯。
1.2 仪器与设备
MTH-012型旋涡混合仪 海门其林贝尔仪器制造有限公司;070-851型PCR仪 德国Biometra公司;DYY-10 C型电泳仪 北京市六一仪器厂;JY04S-3E型凝胶成像分析系统 北京科普尔科技发展有限公司;K5500型超微量核酸测量仪 北京凯奥科技发展有限公司;FP-750荧光分光荧光计 日本分光株式会社。
1.3 方法
1.3.1 细菌基因组提取
取1 mL过夜培养菌液,提取细菌基因组DNA,具体操作方法见细菌基因组DNA提取试剂盒说明书。并用核酸测量仪对其DNA浓度及纯度进行测定。
1.3.2 引物及哑铃环设计
通过Primer Premier 5设计了上游引物(HF)序列:5’-GAAAGAGCAACTGGCCAACG-3’,下游引物(TR)序列:5’-AAAAAAAAAAAAAAAATGCTTGAGCTGATTGCGC-3’。通过DNAMAN设计了可在T4 DNA连接酶的作用下连接成稳定哑铃型的发夹(CP)序列:5’-Phosphate-ATGAACAGG TCTAAAAAAAAAAAAAAACCCAACCCGCCCTA CCCTTTCCTGTTC-3’。并合成了15个T的环外引物5’-TTTTTTTTTTTTTTT-3’,验证实验的可行性。
1.3.3 PCR条件优化
以沙门氏菌基因组DNA为模板,采用HF与TR通过PCR扩增目标序列,获得PCR产物触发后续RCA扩增。为确保得到大量包含15个碱基T(15T)的PCR产物且控制后续诱发RCA反应的“引物”量,达到相对定量效果,本实验对初始引物浓度进行优化,设置了1、2、3、4 μmol/L的梯度变化,在相同的浓度添加量及模板浓度下,寻求最佳的引物初始浓度,最终确定PCR体系为:25 μL 2×EsMaster Mix(Dye),2 μL HF(3 μmol/L),2 μL TR(3 μmol/L),2 μL各浓度基因组DNA,ddHO补至50 μL。
本实验在TR的5’端额外标记了15个A碱基,目的是在PCR结束后获得大量包含15个T的PCR产物。这就造成了引物对的不等长,从而设置了54、56、58、60、62、64 ℃退火温度梯度优化,最终确定PCR程序为:95 ℃预变性5 min,95 ℃变性30 s,56 ℃退火30 s,72 ℃延伸10 s,72 ℃延伸5 min,28个循环。PCR产物采用2%的琼脂糖凝胶电泳验证(140 V、40 min),后利用凝胶成像系统进行观察分析,并按照DNA纯化回收试剂盒步骤纯化PCR产物。
1.3.4 哑铃环状DNA的制备
借助T4 DNA连接酶能封闭DNA链上切口的作用,将所设计的哑铃环状DNA发夹前体的黏性末端连接。并对发夹DNA的反应终浓度及T4 DNA连接酶的加入量进行优化,保障发夹DNA连接成哑铃环的效率。发夹DNA(10 μmol/L)终浓度梯度设置为1、0.5、0.25、0.1 μmol/L,T4 DNA连接酶(350 U/μL)用量梯度设置为0.5、1.0、1.5、2、2.5 μL。最终确定连接反应为5 μL发夹DNA、10 μL 10×T4 DNA连接缓冲液和83 μL ddHO混合均匀后95 ℃保温5 min后37 ℃孵育30 min,使发夹DNA变性,充分进行碱基互补配对。后加入2 μL T4 DNA连接酶,37 ℃孵育3 h,65 ℃灭酶10 min,所制得的连接产物可直接使用或-20 ℃保存备用。
将连接后的哑铃环状DNA产物20 μL加入loading buffer染色后在15%的聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,PAGE)表征(140 V,1 h),后用3×Gel Red染色液(45 mL蒸馏水,5 mL 1 mol/L的NaCl溶液,15 μL 10 000×Gel Red母液)室温避光振荡染色30 min。最终利用凝胶成像系统进行观察分析。
1.3.5 RCA扩增及G-四链体荧光检测的条件及优化
RCA扩增是借助2.0 DNA聚合酶强烈的链置换能力实现的,为达到快速检测的目的,对哑铃环状DNA用量、RCA反应时间及温度进行优化。哑铃环状DNA(0.5 μmol/L)的加入终浓度设置为50、100、150、200、250、300 nmol/L;反应时间设置为20、40、60、80、100 min;反应温度梯度设置为56、59、62、65、68 ℃。因RCA扩增产物为冗长且包含富G序列的ssDNA,为判断该产物是否在扩增结束后自身形成二级结构,对ThT诱导折叠G-四链体结构造成干扰,因此将RCA扩增产物与ThT混匀后95 ℃变性5 min后4 ℃孵育5 min处理与直接室温孵育对比,并对ThT用量及孵育时间进行优化,将ThT(1 mmol/L)浓度梯度设置为5、10、20、30、40、50 nmol/L;孵育时间梯度设置为30、40、50、60、70 min。
最终确定反应条件为20 μL哑铃环状DNA,10 μL PCR纯化产物,5 μL 10×2.0 DNA聚合酶缓冲液,4 μL 2.5 mmol/L dNTP Mix溶液,2.5 μL 100 mmol/L MgSO溶液,7.5 μL ddHO混合均匀后95 ℃变性5 min后4 ℃孵育5 min,加入1 μL 10×2.0 DNA聚合酶,59 ℃孵育1 h。在RCA扩增产物中加入1.5 μL ThT溶液,95 ℃变性5 min,4 ℃孵育5 min后室温孵育1 h,最终将50 μL反应液转移到狭缝比色皿中,用荧光分光光度计测定激发波长425 nm、发射波长486 nm下的荧光值,选择最优条件用于后续实验。
1.3.6 基于G-四链体的RCA反应可行性验证
为了排除实验结果的假阳性和验证哑铃环状DNA的连接效果,以合成的15个T的靶标序列为模板进行RCA扩增G-四链体的可行性验证。根据1.3.5节反应条件对2 μL合成靶标进行阳性对照,无靶标的阴性对照,无2.0 DNA聚合酶对照和无哑铃环状DNA模板对照。最终测量激发波长425 nm、发射波长450~550 nm的荧光信号,同时取5 μL扩增产物采用1%的琼脂糖凝胶电泳验证结果,判定RCA反应扩增G-四链体的信号放大策略的可行性。
1.3.7 基于G-四链体的PCR-RCA方法检测沙门氏菌
1.3.7.1 方法特异性
对过夜培养的革兰氏阴性菌(大肠埃希氏菌、志贺氏菌、铜绿假单胞菌、甲型副伤寒沙门氏菌、丙型副伤寒沙门氏菌和鼠伤寒沙门氏菌)和革兰氏阳性菌(单核细胞性李斯特菌和金黄色葡萄球菌)的基因组DNA进行提取。根据实验优化的最佳条件进行基于G-四链体的PCR-RCA方法检测,测定486 nm下的荧光信号,验证构建方法对沙门氏菌的特异性。
1.3.7.2 方法灵敏度
取过夜培养的沙门氏菌菌液1 mL进行基因组DNA提取,并将提取的核酸进行梯度稀释,测其核酸含量,按照优化好的条件进行基于G-四链体的PCR-RCA方法检测,测量发射波长为450~550 nm的荧光信号,并取486 nm的荧光值,判断此方法的灵敏度。
1.3.7.3 方法检出限(limit of detection,LOD)
取培养的沙门氏菌菌液,用0.85%的生理盐水进行10 倍梯度稀释,每个稀释度取1 mL进行菌落计数。将上述各稀释度菌液5 mL接种于45 mL灭菌牛奶中,均质后取1 mL提取基因组DNA,对基于G-四链体的PCR-RCA方法进行牛奶加标样品中LOD分析。根据文献[28]计算方法:LOD=3/(为空白样品的标准偏差,为校准曲线的斜率),确定此方法的LOD。
1.4 数据处理
相关数据的处理及标准曲线的计算采用Microsoft Excel 2010软件,图表的绘制采用GraphPad Prism 8软件,并采用Adobe Photoshop CS5软件对图片进行了组合排列。
2 结果与分析
2.1 PCR条件优化
引物与模板相互作用,在相同模板浓度下,PCR产物会随着引物浓度的增加而增多,但引物浓度过大又会造成模板经指数扩增后的产物无区别,干扰后续的定量效果,因此进行引物浓度优化。图1显示,当引物浓度达3 μmol/L后,LOD为0.17 pg/μL,且不随着引物浓度增加而变化,因此选用引物浓度3 μmol/L用于后续实验。
因TR的5’端引入15个碱基A,导致上下游引物不等长,值相差较大,因此进行退火温度优化。图1E为退火温度依次为54、56、58、60、62、64 ℃时的琼脂糖凝胶电泳结果,当退火温度为56 ℃时,产物较亮,且无非特异扩增,因此选取56 ℃用于后续实验。
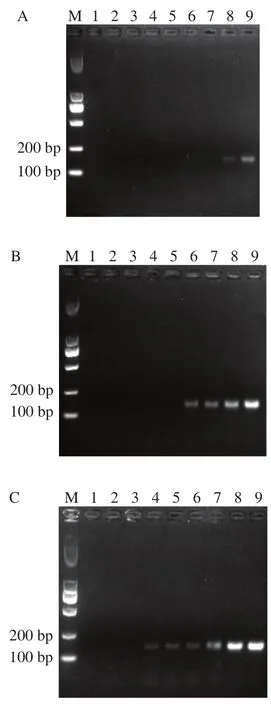
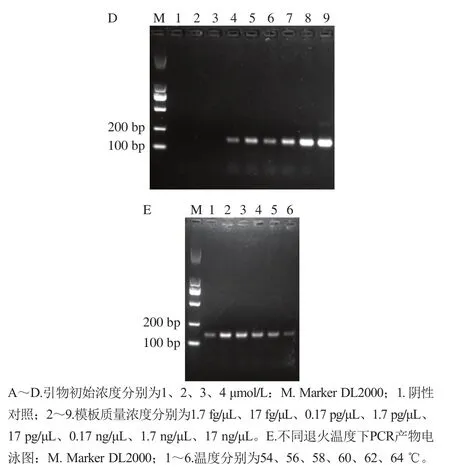
图1 PCR条件的优化Fig. 1 Optimization of PCR conditions
2.2 发夹DNA的连接及优化
RCA反应的关键是构建一个完整的环状模板,而本实验根据G-四链体的反向互补序列及PCR扩增3’端的15个T碱基产物,设计出自身可折叠成半哑铃结构的发夹DNA,且2个发夹DNA的黏性末端又可被T4 DNA连接酶连接成封闭的哑铃环。图2A~D分别是发夹DNA加入的终浓度1、0.5、0.25、0.1 μmol/L,T4 DNA连接酶添加量0.5、1、1.5、2、2.5 μL的连接产物。不难发现发夹探针浓度相对较低时,整个体系的连接效率变高,但也依然存在未连接的原始探针。因此为保障连接的哑铃环状DNA产物量的相对较多,又避免未连接探针的绝对浪费,选取探针终浓度为0.5 μmol/L,T4 DNA连接酶量为2 μL的连接产物用于后续实验。
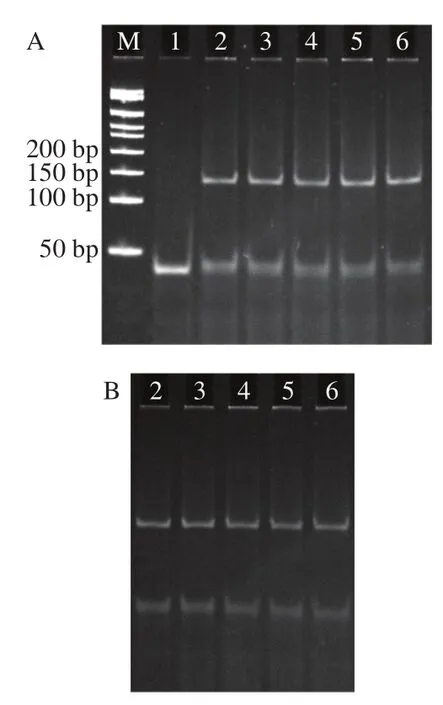

图2 发夹DNA的连接优化Fig. 2 Optimization of hairpin DNA ligation
2.3 RCA扩增及G-四链体荧光检测的条件及优化
2.3.1 RCA反应条件优化
RCA反应是扩增G-四链体从而实现信号放大的关键因素,对后续检测的灵敏度起着重要作用,借助2.0 DNA聚合酶具有的5’→3’聚合酶活性和强链置换活性,可实现高效的等温扩增。RCA反应时间及温度均会对2.0 DNA聚合酶的效率造成干扰,从而影响RCA扩增的产物量,而哑铃环状DNA用量是决定扩增产物多少的关键因素,哑铃环较多,会造成模板浪费,反之会影响实验的灵敏度。
由图3A可见,随着哑铃环状DNA浓度从50 nmol/L增加到200 nmol/L,荧光强度逐渐加强到最大,而超过200 nmol/L时,可能因添加的浓度过大,导致非特异扩增增强,从而富G产物积累减少,荧光值降低。因此选择哑铃环状DNA加入量为200 nmol/L用于后续实验。由图3B可见,随着反应时间从20 min增加到60 min,荧光信号逐渐增强,当时间达到60 min后,荧光值几乎不发生变化,推测此时已达到反应最大限度,因此选择该时间用于后续实验。由图3C可见,随着温度在56~68 ℃的变化,荧光值呈现先升高后下降趋势,推测随着温度的升高,逐渐接近酶的最适温度,此时反应效率最大,但当温度持续上升,使聚合酶在高温下部分失活,又环外引物受值影响互补配对能力下降,故荧光信号降低,因此选择59 ℃进行后续实验。
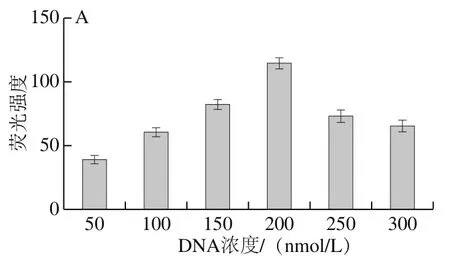
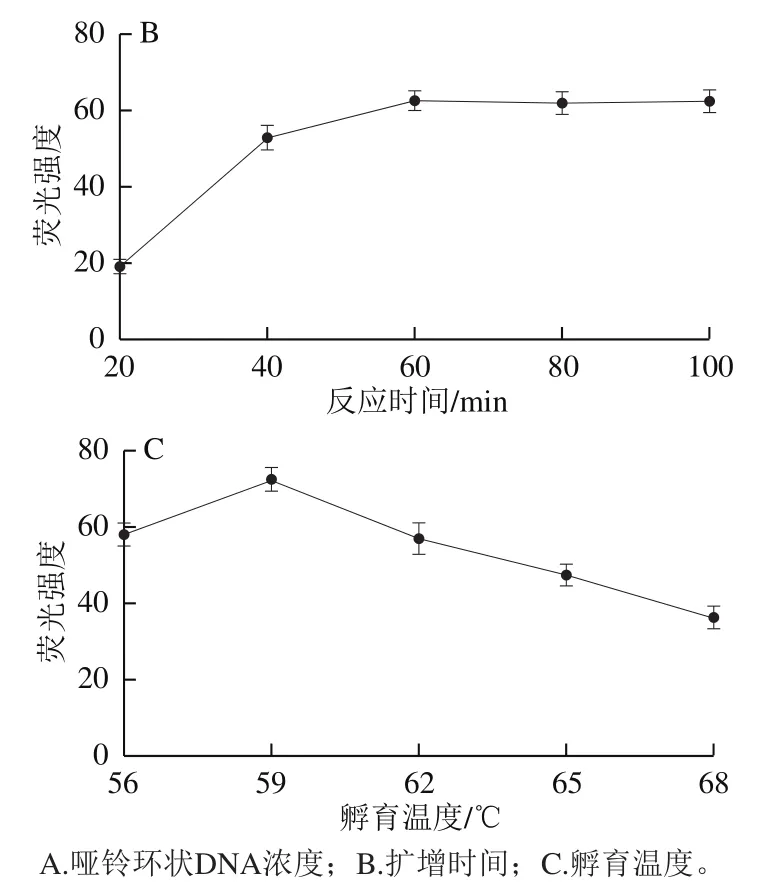
图3 RCA反应条件优化Fig. 3 Optimization of RCA reaction conditions
2.3.2 G-四链体荧光检测条件优化
ThT的旋转结构致使自身荧光值低,但当其特异性识别G-四链体并嵌入其中时,会产生强烈的荧光信号,从而达到检测的目的。因此G-四链体结构形成的多少,ThT的用量及二者相互作用的时间都会对实验的灵敏度有较大影响。

图4G-四链体荧光检测条件优化Fig. 4 Optimization of fluorescence detection conditions for G-quadruplex
图4A为对RCA扩增的长链ssDNA产物进行95 ℃、5 min,4 ℃、5 min处理和直接室温孵育的检测结果,发现RCA产物与ThT经过热变性处理,荧光信号得到明显提升。推测是长链ssDNA自身折叠成二级结构,使富G序列被包裹,从而影响了ThT诱导G-四链体结构的功能。因此,选择热变性处理后再进行室温孵育的方法进行后续实验。图4B为ThT终浓度由5~50 μmol/L的优化结果,当浓度从5~30 μmol/L时,荧光值逐渐增大且达到最大,但浓度超过30 μmol/L,荧光值呈现降低趋势,推测是因溶解ThT粉末的溶液为50%的冰乙酸,自身pH值较低,在过酸的溶液中G-四链体结构形成受到影响所致。因此,选择30 μmol/L的ThT添加量作为后续条件。图4C为RCA产物与ThT室温孵育时间优化结果,当二者作用60 min时,荧光信号达到最大值,且随着时间的延长,荧光值基本不再有大幅度的变化,因此选择60 min孵育时间作为后续条件。
2.4 基于G-四链体的RCA反应可行性验证
通过通用引物对沙门氏菌的基因组DNA进行PCR扩增,得到3’端含有15个T的PCR产物,将其变性解旋后,再诱发RCA反应和ThT荧光检测,因此15个T碱基能否引发RCA反应是实验的关键。同时也针对RCA反应的发生条件,验证实验是否存在假阳性和哑铃环状DNA的连接效果,进行了如下对照。
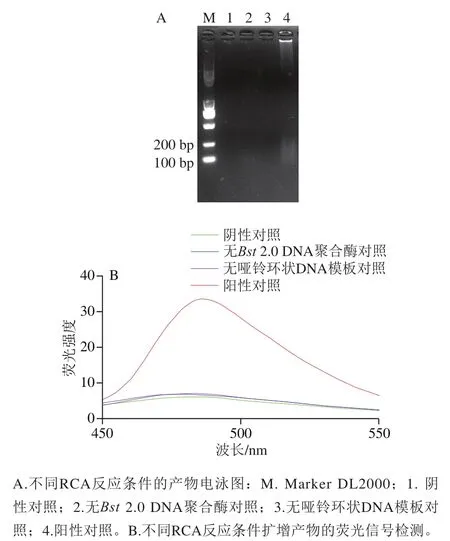
图5 基于G-四链体的RCA反应可行性验证Fig. 5 Feasibility verification of G-quadruplex based RCA reaction
对比琼脂糖凝胶电泳图(图5A)与荧光信号(图5B)可知,阴性对照、无2.0 DNA聚合酶对照和无哑铃环状DNA模板的对照反应体系,均不能诱发RCA反应,故也不能产生强烈的荧光信号,故本实验构建的基于G-四链体的RCA技术检测沙门氏菌的方法具有良好的可行性。
2.5 基于G-四链体的PCR-RCA方法检测沙门氏菌的特异性
为评价方法的特异性,对1.1节中菌株进行基因组DNA提取,根据优化实验得到的最佳反应条件进行基于G-四链体的PCR-RCA扩增及ThT诱导产物增强自身荧光强度的检测。在保证其他菌株的DNA浓度大于沙门氏菌DNA浓度和其他反应条件相同情况下,验证构建方法的特异性。由图6可知,含目标DNA的反应体系产生了明显的荧光信号,而其他非目标菌的荧光信号与空白对照相差无几,证明基于G-四链体的PCR-RCA方法对沙门氏菌具有高度的特异性。

图6 基于G-四链体的PCR-RCA检测方法的特异性Fig. 6 Specificity of G-quadruplex-based PCR-RCA detection method
2.6 基于G-四链体的PCR-RCA方法检测沙门氏菌的灵敏度
取1 mL过夜培养的沙门氏菌,提取基因组DNA后进行10 倍系列稀释,得到1.7 fg/μL、17 fg/μL、0.17 pg/μL、1.7 pg/μL、17 pg/μL、0.17 ng/μL、1.7 ng/μL、17 ng/μL的靶标样品,在优化的反应条件下进行基于G-四链体的PCR-RCA方法的检测,以确定该方法的灵敏度。由图7A可知,在17 fg/μL~1.7 ng/μL的基因组DNA模板下,随着靶标质量浓度的逐渐增加,荧光信号也呈现逐渐增强的趋势。且由图7B可知,基因组DNA浓度对数与荧光信号呈现出良好的线性关系,线性回归方程为=2.101+2.872 3(=0.992 5),灵敏度为17 fg/μL,说明构建的基于G-四链体的PCR-RCA检测方法可以对沙门氏菌进行有效的鉴别和定量检测。
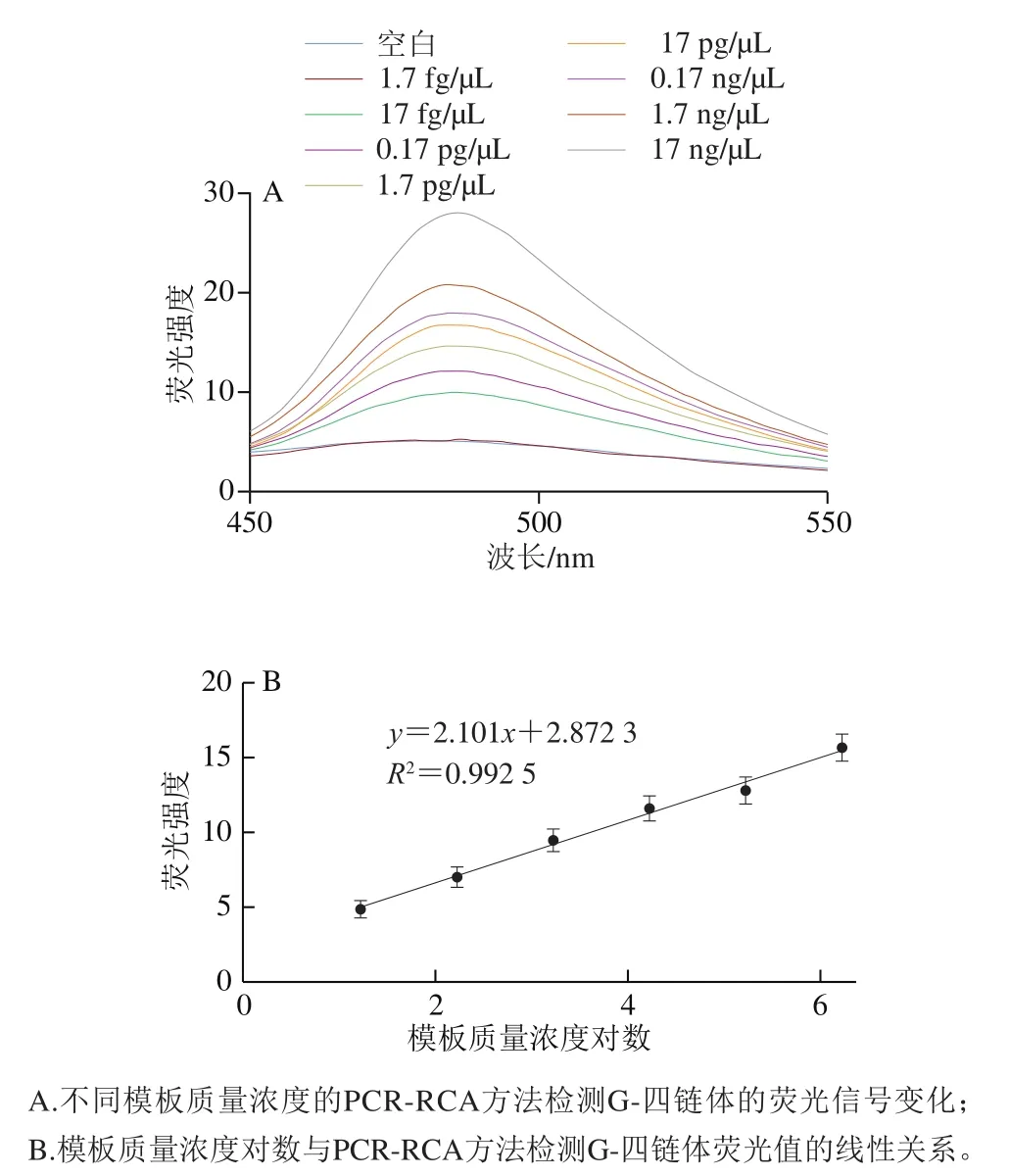
图7 基于G-四链体的PCR-RCA检测方法的灵敏度Fig. 7 Sensitivity of G-quadruplex-based PCR-RCA detection method
2.7 牛奶加标样品中基于G-四链体的PCR-RCA方法的LOD
利用PCR-RCA方法扩增G-四链体荧光检测沙门氏菌的最佳条件,对沙门氏菌标准菌株污染的牛奶进行检测,其中人为污染浓度为10~10CFU/mL,通过观察ThT诱导扩增产物并特异性结合G-四链体的荧光增强效果及对比加标样品污染浓度,进行检测灵敏度计算。由图8A可知,荧光信号随着样品污染程度的增加而增加。且由图8B可知,在10~10CFU/mL样品污染浓度下,其对数与荧光值呈现出良好的梯度变化,计算检测限为4.48 CFU/mL。因此本实验构建的方法在沙门氏菌实际检测中实用性良好,在一定范围内可用于沙门氏菌的定量检测。

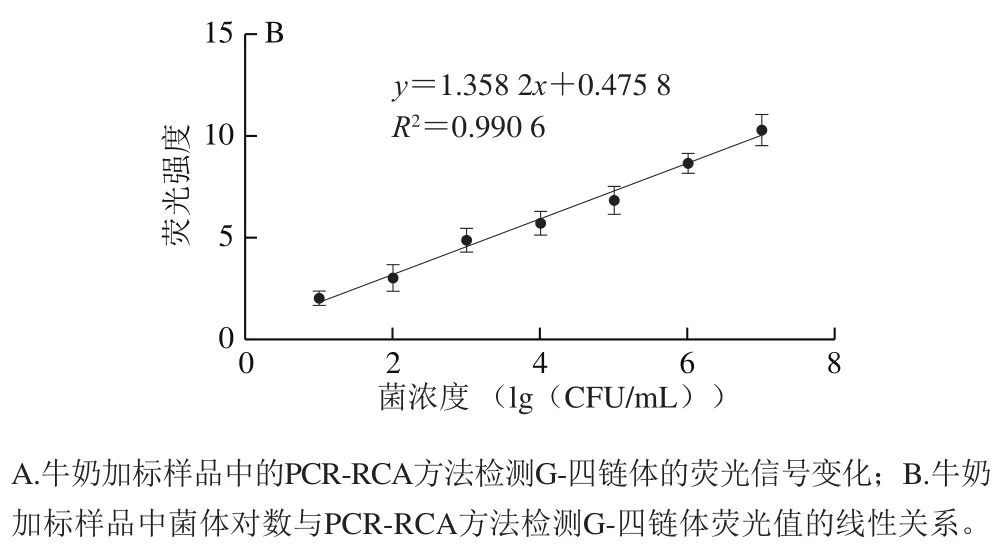
图8 基于G-四链体的PCR-RCA方法检测人工污染沙门氏菌的牛奶样品Fig. 8 Detection of Salmonella in artificially contaminated milk samples by the G-quadruplex-based PCR-RCA method
3 结论与讨论
目前,G-四链体偶联RCA的方法,已实现单核细胞性李斯特菌、克氏杆菌属、microRNA等的检测。但其研究或采用较为复杂的引发RCA扩增的手段,或设计更为复杂的环状模板,增加了实验的难度。本实验建立的基于G-四链体的PCR-RCA双重扩增检测沙门氏菌的方法,特点与优势在于:1)借助PCR扩增,使产物3’端生成的15个T碱基触发后续实验,既可确保引物与靶基因片段识别检测的特异性,又可在核酸第1次扩增时积累大量“引物”,确保实验的灵敏度。该方法实现了对低丰度目标序列的初步扩增,获得与扩增产物等量的“15T”,为后续含“15A”的颈环结构提供RCA扩增引物,通过诱发RCA扩增间接完成对目标序列的检测,也通过RCA扩增得到巨量G-四链体实现荧光信号的分析。因此,该方法对低丰度DNA样品具有应用潜力。2)以“15A”碱基为“桥梁”,可扩展到对不同目标核酸序列的检测中,即只需要根据目标序列设计5’端带“15A”的引物,无需重新设计哑铃环状DNA模板,就可实现对不同目标序列的检测,使方法具备通用性。3)设计带有“AT”两个碱基黏性末端的发夹结构,在T4 DNA连接酶的作用下可产生类似T-载体的连接效果,巧妙高效地连接成哑铃环状DNA模板。此方法有别于其它连接方式,不需要额外添加成环引物和核酸外切酶消化未连接的环,有效减少实验假阳性。4)与荧光定量PCR检测中的荧光染料法相比,本实验方法可弥补由于嵌入核酸的荧光染料无法区分特异性产物与非特异产物造成的检测结果误差大的缺点;与探针法相比,可避免探针设计相对复杂、标记荧光淬灭基团成本高的弊端。最终利用ThT对核酸第2次扩增产物中的重复富G单元特异识别,将核酸信号辅助放大为荧光信号,借助荧光分光光度计即可实现检测的精准定量效果,虽然步骤上略显繁琐,但是其应用范围较荧光定量PCR更为广泛。
G-四链体作为一种高稳定性、广泛存在的功能核酸,以易于修饰和合成、检测方式灵活的优异特征,已应用到多种核酸检测技术中。但如何巧妙地将G-四链体与所检核酸相偶联,积累大量富G序列是其关键。因此进行如下改进:1)PCR扩增的引物浓度是后续生成诱发RCA反应的前提。通过PCR扩增所得的RCA引物浓度高时,虽然灵敏度得到显著提高,但易掩盖RCA引物间的浓度差,直接影响RCA富G产物检测的梯度。因此,本实验优化选择3 μmol/L的PCR引物,刚好能获得最佳RCA扩增梯度。2)与扩增靶标得到5’-磷酸化产物并经过核酸外切酶消化和不对称拖尾PCR (AT-PCR)获得ssDNA产物触发后续RCA反应的方法相比,本实验通过直接对PCR纯化产物进行热变性处理,使解链的3’端15个T碱基与哑铃环状DNA模板相互识别配对,通过链替换扩增积累大量RCA产物,使整个实验操作变得便捷。3)RCA扩增产物为冗长的ssDNA,其自身二级结构会包裹大量重复的富G序列而影响G-四链体结构的形成,因此在进行ThT荧光信号放大检测前,将RCA产物进行热变性及冷却孵育处理,使富G碱基裸露,即可显著提升检测灵敏度。
综上,通过一系列的实验设计与优化,建立了PCRRCA双重核酸扩增与G-四链体核酸辅助检测信号放大联合检测沙门氏菌的新方法,该方法有效性强、稳定性好、灵敏度高,借助荧光分光光度计对所检样品的荧光值进行分析,即实现一定范围内的定量检测。可广泛用于其他食源性致病菌的检测,通用性强,只需改变PCR扩增的引物,即可达到对靶物质的测定。但相比其他信号放大技术,G-四链体/ThT的荧光信号强度易受到缓冲体系中一价阳离子浓度的影响,因此在检测复杂样品和低浓度样品时存在一定的局限性。
