Ag/Co-OMS-2催化氧化邻二甲苯性能研究
2022-07-05颜文秀宋鑫鑫邢一帆吴银素
颜文秀, 宋鑫鑫, 邢一帆, 吴银素
(河北师范大学 化学与材料科学学院,河北 石家庄 050024)
苯系物(苯、甲苯、二甲苯等)常用作各种胶油漆涂料和防水材料的溶剂或稀释剂[1].挥发到大气中的苯系污染物会对人类的生产和生活造成很大危害,因此,苯系污染物的处理迫在眉睫[2-10].催化氧化燃烧法,可以将污染物在催化剂的作用下直接转化为CO2和H2O,是消除这类挥发性有机化合物(VOCs)的高效方法之一[11-17].催化氧化燃烧法的关键是催化剂的选择.
Ⅱ-型氧化锰八面体分子筛(OMS-2)属于α-MnO2的一种.OMS-2具有多变的价态、疏水性以及环境友好等特点,被认为是最有前途的过渡金属氧化物催化剂之一[18-19].在过渡金属氧化物中,钴氧化物在催化燃烧VOCs方面表现出优异的性能[20-22].氧化钴或氧化锰都可以作为主要成分来提高钴锰催化剂的催化氧化能力[23].钴离子与锰离子半径相似,通常将钴离子掺杂到MnO2中或将锰离子掺杂到钴氧化物中,可增强二者对VOCs的催化性能.Zhang等[24]通过草酸盐共沉淀法制备的Co2Mn1Ox催化剂完全催化氧化苯的能力最佳.其高活性的机理可解释为锰取代了Co3O4中的钴,产生晶格缺陷,使催化剂具有高氧迁移率,产生更多的活性氧物种参与反应.Liu等[25]采用溶剂热醇解法制备的5Co1Mn催化剂具有结构缺陷、介孔中空形貌和高比表面积,促进活性氧物种的释放和更多表面活性位点的暴露,从而加速反应物的吸附和转化.Han等[26]采用模板法、浸渍法和酸处理法制备了两相8.8Co3O4-MnO2催化剂.Co2+或Mn2+含量的增加意味着氧空位量的增加,氧空位处激活氧分子而产生的吸附氧物种有助于提高催化剂的催化氧化能力.
上述研究表明Co和Mn协同作用提升了催化剂氧空位或晶格氧的活性,取得了较好的结果,但催化性能有待进一步提高.近期研究发现,Ag具有促进分子氧活化的能力.Deng等[27]制备的Ag-MnOx-H催化剂中,银的掺入可以活化晶格氧和气态O2,产生更多的活性氧物种,提高了催化剂催化氧化苯的能力.银掺杂型催化剂可以通过氧化还原法、水热法、浸渍法等方法制得.银物种可以弱化锰氧键、促进活性氧物种的生成[28],但多数方法只是将银物种均匀地掺杂到催化剂表面,很难达到分子水平掺杂,所以探究实现这一目的的方法很有必要.本文中,笔者以高锰酸钾、硫酸锰、硝酸钴为原料,采用氧化还原共沉淀法得到Co-OMS-2催化剂,再以AgNO3为反应物,利用沉积沉淀法制备Ag/Co-OMS-2催化剂,用于邻二甲苯的催化氧化,并探究了Ag/Co-OMS-2催化氧化邻二甲苯的构效关系.
1 实验部分
1.1 催化剂的制备
1) Co-OMS-2的制备.将一定量的MnSO4和Co(NO3)2溶液混合,滴入过量10 %KMnO4溶液(质量分数)中,并用98 %H2SO4溶液(质量分数)调节体系pH=3.5±0.3.室温搅拌2 h后转入100 mL不锈钢高压反应釜中,120 ℃反应24 h.降至室温,将产品过滤,用去离子水洗至中性,110 ℃干燥12 h,350 ℃焙烧4 h,得到的催化剂记作Co-OMS-2.
2) Ag/Co-OMS-2的制备.Co-OMS-2中加适量蒸馏水形成悬浮液,将其加入AgNO3溶液中,用0.2 mol/L KOH溶液调节体系pH=10,超声2 h后再陈化12 h,将产品过滤并水洗至中性.110 ℃干燥12 h,400 ℃焙烧4 h,得到的催化剂记作Ag/Co-OMS-2.
3) OMS-2的制备.采用与Co-OMS-2相同的制备方法,不加Co(NO3)2溶液,得到OMS-2催化剂.
1.2 催化剂的表征
采用德国Bruker公司的AXS-D8-ADVANCE型XRD确定催化剂的晶相,采用Cu-Kα射线,管电压与管电流分别为40 kV和40 mA,扫描范围10 °~80 °,扫描速度0.2 °/s,步长0.02 °.采用Quantachrome NOVA 4000e型仪器进行N2吸附-解吸测试.120 ℃真空处理4 h,在液氮-196 ℃条件下进行测试.采用Hitachi S-4800扫描电子显微镜(SEM)和能谱分析仪(EDX)分析催化剂的形貌和化学组成.采用FTR-8900傅里叶红外光谱仪和TANFO 3-Desktop拉曼光谱仪分析催化剂的精细结构及晶体缺陷.利用MS-5000电子顺磁共振光谱仪测试催化剂的氧空位含量.
利用Builder PCA-1200化学吸附仪测试催化剂的性能.催化剂用量均为40 mg,升温速率均为10 ℃/min.氢气程序升温吸脱附反应(H2-TPR):催化剂在300 ℃,Ar氛围中处理1 h,再在体积分数为10 % H2/Ar氛围中升温至600 ℃.氧气程序升温吸脱附反应(O2-TPD):催化剂在300 ℃,Ar氛围中处理1 h,再在500 ℃吸氧1 h,降至室温,He氛围中升温至900 ℃.程序升温还原再氧化反应(TPR-TPO):H2-TPR测试,催化剂在体积分数为5 % O2/He氛围中升温至600 ℃[29].CO2程序升温吸脱附反应(CO2-TPD):催化剂在300 ℃,Ar氛围中处理1 h,在100 ℃吸CO21 h,然后在He氛围中升温至400 ℃.NH3程序升温吸脱附反应(NH3-TPD):将CO2-TPD中的CO2吸附变为NH3吸附,其余步骤相同.
1.3 催化剂的活性测试
催化剂的性能评价在常压固定床石英管反应器中进行,约0.5 g催化剂和1.5 g石英砂(均为3~4.5×105nm)混合后置于石英管中央,将体积分数为0.05 %的邻二甲苯与4∶1的氮氧混合气体通入反应器,在石英管内发生气固反应,通过FID检测器与填充柱、转化炉检测催化氧化反应后CO2的浓度.
催化剂活性测试条件:φ(邻二甲苯)= 0.05 %;体积分数为20 %的O2/N2平衡;总流速为50 mL/min,W/F(催化剂用量/反应气体流量)=0.60 g/(mL·s-1).
2 结果与讨论
2.1 催化剂的活性测试
催化剂的活性测试结果见图1a.可见,OMS-2对邻二甲苯的催化活性最差,Co-OMS-2的活性明显提高.Ag/Co-OMS-2表现出最好的催化性能,其T100,T50,T20分别为180,162,152 ℃(T100,T50,T20分别对应CO2产率为100 %,50 %,20 %的温度),较OMS-2分别降低了30,32,35 ℃.Ag/Co-OMS-2的催化活性较笔者前期研究的Ag-OMS-2的活性也有明显提升[29].这些结果表明,Ag和Co之间具有协同作用.图1b为Ag/Co-OMS-2在180 ℃ 下连续催化氧化邻二甲苯60 h的稳定性测试结果,其CO2产率为97 %~100 %,说明该催化剂有良好的稳定性.另外,3种催化剂的面积比速率顺序为Ag/Co-OMS-2>Co-OMS-2>OMS-2(见表1),说明Ag和Co掺杂后活性的提高不是由比表面积增大引起的.

图1 3种催化剂的催化活性(a)及Ag/Co-OMS-2的稳定性(b)Fig.1 Catalytic Activity of Three Catalysts(a) and Stability of Ag/Co-OMS-2(b)

图2 3种催化剂的阿伦尼乌斯图Fig.2 Arrhenius Plots of Three Catalysts for o-xylene Oxidation
使用一阶Arrhenius方程计算3种催化剂催化氧化邻二甲苯的表观活化能(Ea).计算公式:r=-kc=-(Aexp(-Ea/RT))c,其中c,r,k和A分别为邻二甲苯体积分数、反应速率(μmol/(g·s))、速率常数(s-1)和指前因子.OMS-2,CO-OMS-2,Ag/CO-OMS-2的温度分别为455,438,423 K,3种催化剂的CO2产率均小于20 %.所得Ea分别为189,129,78 kJ/mol(见图2).3种催化剂Ea值大小顺序为OMS-2>Co-OMS-2>Ag/Co-OMS-2.该结果与图1显示的催化剂活性结果相符,表明Ag和Co掺杂后,显著降低了它的活化能.这可能与它们掺杂后引起了结构和表面组成的改变有关.
2.2 催化剂的结构和表面性质
图3a为催化剂的XRD图.可以看出,OMS-2在12.8 °,18.1 °,28.8 °,37.3 °,41.9 °,49.8 °,56.3 °,60.1 °,65.7 °,69.4 °处出现衍射峰,符合标准图谱JCDPS 44-0141[29];而Co-OMS-2和Ag/Co-OMS-2只在28.8 °,37.6 °,41.9 °,49.8 °,60.1 °,65.7 °处出现弱的衍射峰.逐渐提高焙烧温度至450 ℃后衍射峰增强(图3b).表明Co-OMS-2为微晶态的OMS-2,Ag同样高度分散于Co-OMS-2中,同时,Co-OMS-2和Ag/Co-OMS-2在37.6 °的衍射峰较OMS-2高移了约0.3 °.可能是Co掺入OMS-2晶格中且引起OMS-2的晶格畸变.
由表1中催化剂的比表面积结果发现,与OMS-2相比,Co-OMS-2的比表面积明显增大.掺入银物种后,Ag/Co-OMS-2的比表面积较Co-OMS-2有所降低,但变化不大.进一步说明Co和Ag均匀掺入OMS-2中.
图4是催化剂的SEM和EDX谱图.由图4a,b,c可知,3种催化剂均具有棒状形貌.其中,OMS-2呈长棒状,其长和宽分别为150~200 nm和25~30 nm;而Co-OMS-2和Ag/Co-OMS-2则为团聚的细短棒,其长和宽分别为120~160 nm和20~25 nm.

图3 3种催化剂的XRD图(a)及催化剂在不同焙烧温度的XRD图(b)Fig.3 XRD Patterns of Three Catalysts(a) and Catalysts with Different Calcination Temperatures(b)

表1 3种催化剂的面积比速率及元素组成Tab.1 Area Ratio Rate and Element Composition of Three Catalysts
Co/Mn原子比通过EDX测试获得.
由图4a′,b′,c′可知,OMS-2由Mn,O及K元素组成,且其Mn/O物质的量比(1.03∶2)符合MnO2的化学计量比.与OMS-2相比,Co-OMS-2中增加了Co元素,但同时Mn的含量减少,而K元素含量没有明显变化.其(Mn+Co)/O 物质的量比(1.03∶2) 符合MnO2的化学计量比,说明Co元素替代了部分Mn进入OMS-2的晶格中.与Co-OMS-2相比,Ag/Co-OMS-2在Ag掺入的同时,Mn的含量没有明显变化,但Co的含量减少,说明Ag可能替代了部分Co;同时,其K的含量相对Co-OMS-2和OMS-2增加.这可能是由Co-OMS-2制备Ag/Co-OMS-2过程中使用KOH调节pH引发的结果.
图5a是催化剂的FT-IR图.3 456,1 636 cm-1处的峰归属于O—H 的伸缩和弯曲振动峰.OMS-2在477,538,604,727 cm-1处的峰归属于[MnO6]八面体中Mn—O键的伸缩振动峰[30].与OMS-2相比,Co-OMS-2和Ag/Co-OMS-2只在477,572,727 cm-1处出现弱峰,说明Co或Ag物种掺入OMS-2的晶格中.这一结果与EDX的分析结果一致.
由图5b可见,OMS-2在179,387,575,633 cm-1处出现了锰氧化物的拉曼特征峰.其中179,387 cm-1的峰归属于Mn—O—Mn 的变形振动,576,634 cm-1的峰为α-MnO2四方结构的(2×2)隧道内[MnO6]八面体的Mn—O—Mn拉伸振动[23].掺入Co后,OMS-2的特征峰分别移至190,392,584,646 cm-1,而且上述衍射峰的强度明显弱于OMS-2.Ag/Co-OMS-2的拉曼特征峰与Co-OMS-2类似.这些结果进一步说明Co和Ag物种掺入OMS-2的晶格中,使OMS-2产生了结构缺陷.
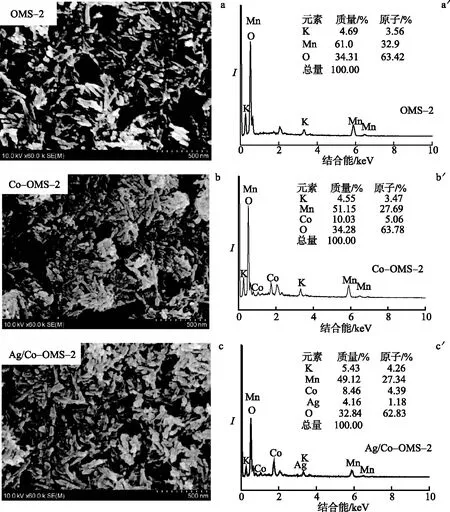
图4 3种催化剂的SEM(a,b,c)和EDX(a′,b′,c′)谱图Fig.4 SEM(a,b,c) and EDX(a′,b′,c′) Profiles of Three Catalysts
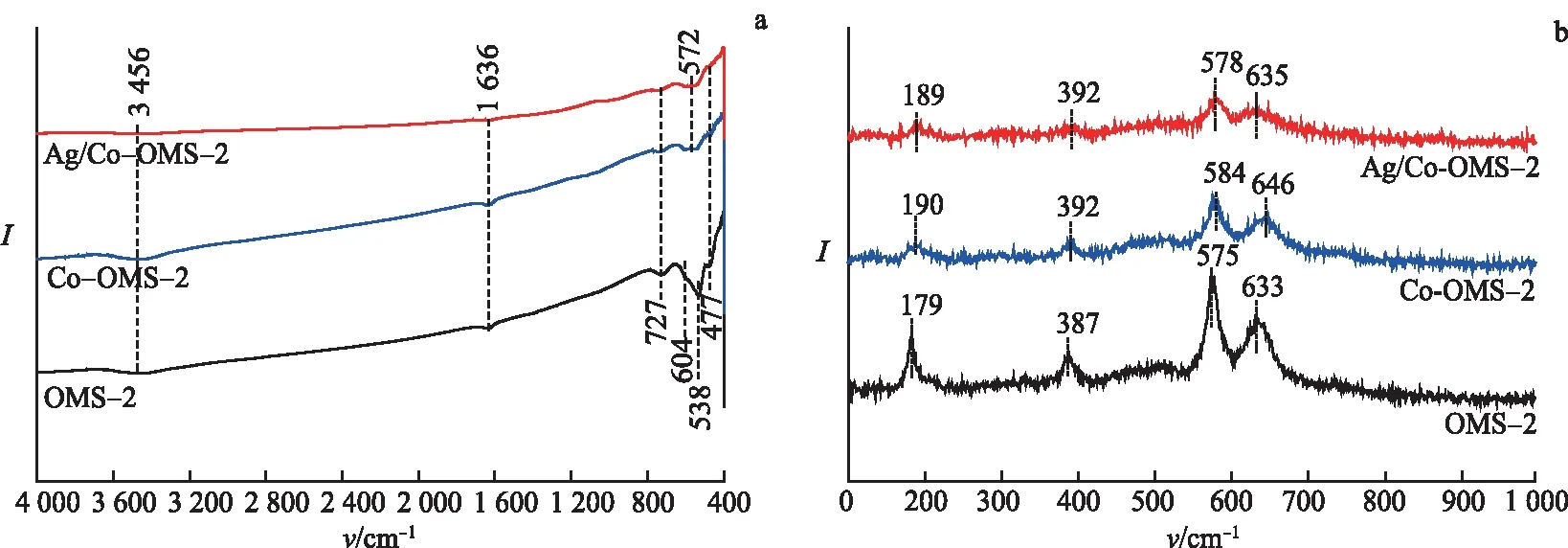
图5 3种催化剂的FT-IR(a)及拉曼图谱(b)Fig.5 FT-IR Spectra(a) and Raman Spectra(b) of Three Catalysts
2.3 催化剂的氧化还原性能及表面酸碱性
图6a为催化剂的H2-TPR测试谱图,用其测定催化剂对H2的氧化能力.由图6a可知,OMS-2在304 ℃处出现1个宽而弱的峰.Co-OMS-2在286,307 ℃出现2个还原峰,分别归属于MnO2到Mn2O3的还原峰和Mn2O3到MnO及Co2O3到CoO混合还原峰[31].Ag掺入后,Ag/Co-OMS-2在中心位置(260 ℃处)出现1个强还原峰,可归属于Olatt(Ag),Olatt(Mn) 和Olatt(Ni)物种的混合还原峰.最低的还原温度预示着其最强的低温还原性[27].
图6b为催化剂的O2-TPD测试谱图,用其测定催化剂氧物种的活化能力.其200~400 ℃峰为表面物理或化学吸附的氧物种的脱附峰,400 ℃以上的峰归属于体相晶格氧的脱附[32].由图6b可知,掺入Co后,表面和体相氧物种的脱附温度明显降低.由FT-IR和拉曼光谱可知,Co掺入OMS-2的晶格中,这可能是其氧物种脱附温度降低的原因.进一步掺杂Ag后表面活性氧物种的还原温度进一步降低,同时,体相晶格氧的脱附峰数量增加.预示着Ag和Co同时掺入OMS-2后改变了体相晶格氧的类型.
图6c为催化剂的TPR-TPO测试谱图,用于确定催化剂被还原后的再氧化能力,即催化剂晶格氧与分子氧的交换能力.由图6c可知,Co(尤其是Ag)掺入OMS-2中,显著降低了分子氧的再氧化温度,即分子氧与晶格氧的交换能力增大.
图6d为催化剂的电子顺磁共振(EPR)信号图.一般情况下,EPR信号是由氧空位捕获的未配对电子产生.EPR信号越强,预示着催化剂所含氧空位越多[24].由图6d可知,3种催化剂在g=2.004时均出现EPR信号,强度顺序为Co-OMS-2>Ag/Co-OMS-2>OMS-2.说明在OMS-2中掺入Co,产生了较多的氧空位.综合H2-TPR,O2-TPD及TPR-TPO的氧化还原性能,进一步说明了3种催化剂中氧空位的差异.
综合上述结构表征和氧化还原性能测试结果,Co取代Mn掺入OMS-2晶格中,引起OMS-2比表面积的增大和晶格畸变,产生了氧空位,同时增加晶格氧的流动性,提升了晶格氧的低温还原能力.Ag的掺入进一步增加了催化剂分子氧与晶格氧的交换能力,这可能是Ag/Co-OMS-2活性高的主要原因.
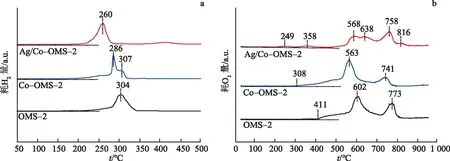
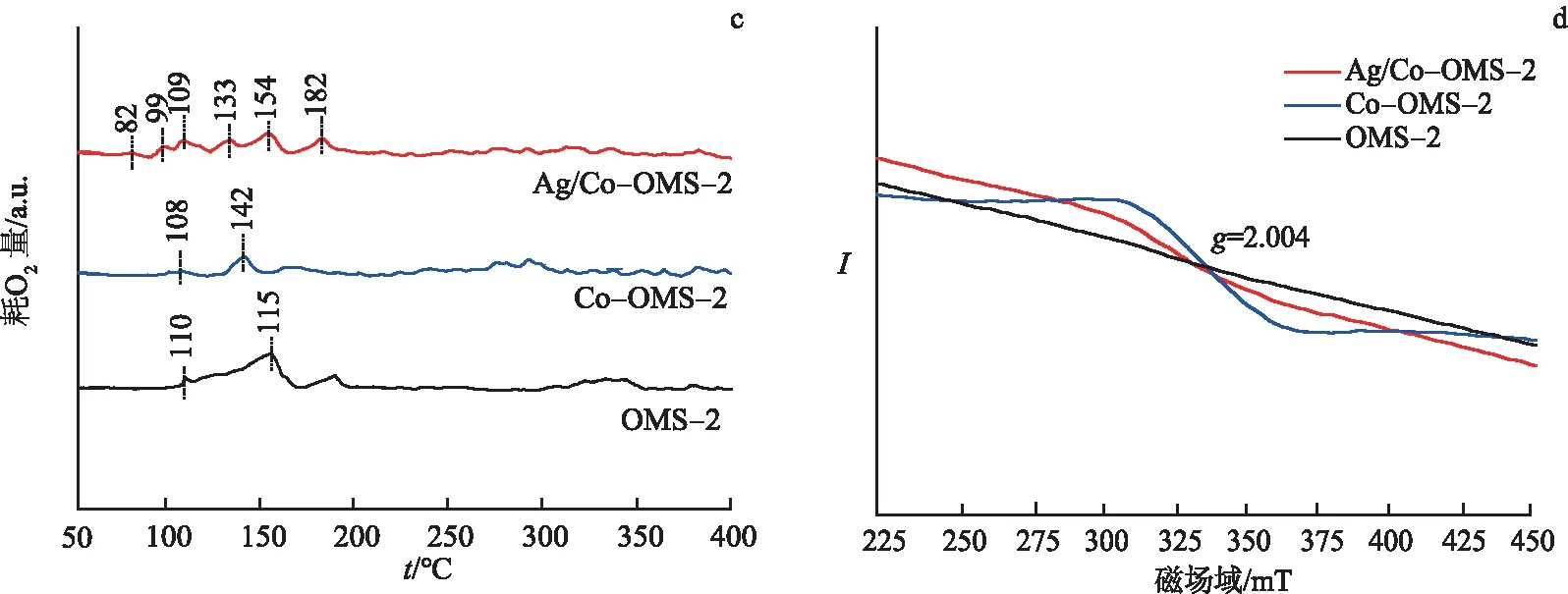
图6 3种催化剂的H2-TPR(a),O2-TPD(b),TPR-TPO(c)及EPR(d)图谱Fig.6 H2-TPR(a),O2-TPD(b),TPR-TPO(c) Profiles and EPR Spectra(d) of Three Catalysts
图7为催化剂的CO2-TPD(a)和NH3-TPD(b)谱图.可知,当Co(尤其是Ag)掺入OMS-2后,其表面酸性增大,而表面碱性降低.相关研究表明,催化剂存在适度的表面酸性有利于苯系物的活化,而较低的表面碱性则可促进苯系物催化氧化的中间物种羧酸盐及最终产物CO2的脱附.这可能也是Co-OMS-2和Ag/Co-OMS-2活性提高的原因之一.
3 结 论
以高锰酸钾、硫酸锰、硝酸钴及AgNO3为反应物,采用氧化还原沉淀法和沉积沉淀法得到了Ag/Co-OMS-2催化剂,并用于邻二甲苯的催化氧化.活性测试结果表明,掺入Co和Ag后,催化剂的活性明显增加.Ag/Co-OMS-2的活性最佳,其T100,T50,T20分别为180,162,152 ℃,较OMS-2的T100,T50,T20分别降低了30,32,35 ℃.各种表征结果表明,Co掺入OMS-2晶格中,使OMS-2发生了晶格畸变,使其氧空位含量增加的同时提升了OMS-2晶格氧的低温还原能力,而Ag的加入进一步提升了分子氧的活化能力;并且Co和Ag的加入增加了OMS-2的表面酸性,减少了其表面碱性,进而促进了邻二甲苯在OMS-2上的吸附能力及其氧化中间物种羧酸盐和最终产物CO2的脱附能力.高的氧空位含量、良好的低温还原性以及增强的分子氧交换能力是Ag/Co-OMS-2催化活性高的原因,且Ag/Co-OMS-2具有良好的稳定性.

图7 3种催化剂的CO2-TPD(a)及NH3-TPD(b)谱图Fig.7 CO2-TPD(a) and NH3-TPD(b) Profiles of Three Catalysts
