紫苏油微胶囊的性质与体外模拟消化研究
2022-06-23马明月姜宏宇
马明月, 姜宏宇, 张 华,2
(延边大学农学院食品与生物科学系1,延吉 133002) (东北寒区肉牛科技创新教育部工程研究中心;延边大学2,延吉 133002)
紫苏油是一种含高度不饱和脂肪酸的天然油脂,PUFA质量分数高达98%,所含主要成份为α-亚麻酸,质量分数高达50%~70%,是目前所发现的所有天然植物油中这种脂肪酸含量最高的[1]。成人摄入脂肪后,在胃中由胃脂肪酶消化5%~30%,在小肠中由胆囊和胰腺分泌物的协调作用消化和吸收30%~75%[2]。为保留其功能特性,降低在胃环境下的损失,将PUFA以结构上和功能上完整的形式递送到小肠再释放出来进行消化吸收,利用微胶囊技术,让壁材对油脂进行包埋,在其表面形成保护层,可以有效避免这些敏感性成分直接与外界的光、氧、水以及酸碱性物质接触,保留PUFA和其他亲脂成分的结构和功能,增强它们的消化,吸收和生理利用能力[3,4]。
制备微胶囊的方法有喷雾干燥和复凝聚法等。喷雾干燥法是将芯材分散在壁材溶液中,在高温气流中将芯材、壁材的混合溶液雾化,使溶液中的溶剂迅速蒸发,最终使壁材固化而得到微胶囊产品[5]。复凝聚法是先将芯材均匀地分散在带相反电荷的混合壁材溶液中,通过调节体系的温度、pH或浓度,使壁材发生静电作用,壁材在相互吸引的过程中,将芯材包裹住,凝聚成微胶囊[6]。复凝聚法必须选用2种或以上带有正负不同电荷的高分子化合物作为复合壁材,当改变溶液体系pH至临界值时,作为两性电解质的蛋白质携带着正电荷,与携带着负电荷的碳水化合物间因静电作用力而形成复合凝聚物,从而将芯材包埋形成微胶囊[7]。相比于其他微胶囊技术如喷雾干燥,它最主要的优点是具有极高的包埋率和缓释性能,并且所需条件温和,不涉及高温。因此,在这个过程中可以避免芯材的活性物质和风味损失或挥发。
海藻酸钠是从海藻中提取的一种天然多糖类化合物,具有良好的稳定性、絮凝性、成膜性、增稠性和螯合性[8]。明胶是一种从动物(猪、牛、马、鱼等)的皮肤、骨骼、筋腱等含胶原蛋白的组织中经过一系列化学处理以后部分水解的多肽和蛋白质的混合物,具有良好的成膜性、乳化性、水溶性、生物相容性和可生物降解能力,并且无毒害、成本低[9,10]。宋双居等[11]以明胶和海藻酸钠为壁材,利用体外释放实验测试微胶囊的释放特性。结果表明:合成的明胶-海藻酸钠微胶囊对啶虫脒有良好的缓释作用。因此,它们是一种制备微胶囊的理想包埋材料。Dong等[12]以明胶-阿拉伯树胶为壁材,转谷氨酰胺酶为固化剂,采用复凝聚法制备薄荷油微胶囊。得知薄荷油微胶囊的释放分别遵循热水一级释放动力学模型和高温烘箱零级释放动力学模型;谭睿等[13]以明胶分别与阿拉伯胶、果胶、羧甲基纤维素钠组合为壁材,采用复凝聚法制备绿咖啡油微胶囊,包埋率达到85%以上;Yang等[14]以壳聚糖为壁材,采用复凝聚法制备香草精油微胶囊。结果表明该微胶囊具有较长的残留作用和较高的热稳定性,可作为一种优质食品香料。
近年来,随着微胶囊技术的成熟,在制备紫苏油微胶囊的工艺研究部分较为集中,但对于紫苏油微胶囊在人体胃肠道内的消化吸收研究较少。因此,本研究通过以明胶和海藻酸钠为壁材,采用复凝聚法制备紫苏油微胶囊,并测定微胶囊的理化特性、抗潮性、耐热性以及在模拟胃肠道条件下的消化吸收能力,为深入研究紫苏油微胶囊在胃肠道内的消化吸收能力提供依据。
1 材料与方法
1.1 材料与试剂
紫苏油、海藻酸钠、明胶B型、转谷氨酰胺酶,均为食品级;胃蛋白酶(猪源)、胰蛋白酶。
1.2 仪器与设备
FK-FSH-2A型高速分散均质机,FD-1C-50型真空冷冻干燥机,DK-2型可调式电沙浴,DCY-SY12型恒温水浴氮吹仪,HWS-50B型恒温恒湿箱,GC-2010plus型气相色谱仪。
1.3 方法
1.3.1 紫苏油微胶囊的制备
分别制备质量分数为2%的明胶和海藻酸钠溶液(明胶与海藻酸钠的质量比为4∶1[15]),先取8 mL的紫苏油加入海藻酸钠溶液,再加入0.04 mL的乳化剂司班80,开始进行第1次均质乳化,然后加入明胶溶液进行第2次均质乳化得到水包油乳液,2次均质速度均为10 000 r/min。使用10%的冰乙酸溶液调节乳液pH为4.25时进行复凝聚反应,反应条件为45 ℃、600 r/min,9 min。复凝聚反应结束后将体系降温至10 ℃以下维持20 min进行凝胶,每5 min搅拌1次。凝胶结束后,去除上清液并加入去离子水洗涤,再用10%氢氧化钠溶液逐滴加入调节pH为6,加入1.28 g的转谷氨酰胺酶(TG酶)低温固化处理,固化温度为15 ℃,固化时间为2 h。固化结束后,经过真空冷冻干燥得到微胶囊。
真空冷冻干燥:将所得的微胶囊液倒入冻干物料盘中,厚度不超过1 cm,放入-80 ℃冰箱中预冷12 h后经真空冷冻干燥机(-50 ℃、1 Pa)冷冻干燥得到紫苏油微胶囊产品。
1.3.2 紫苏油微胶囊的含水量测定
准确称取3 g微胶囊样品,在105 ℃的烘箱中干燥3 h,取出置于干燥器中冷却称重。重复上述干燥操作,直到最后2次所测的样品质量差小于1 mg,所得到的称重前后微胶囊产品的质量差即为含水量。每一样品进行3次平行实验。
1.3.3 紫苏油微胶囊的堆积密度测定
准确称取3 g微胶囊样品,缓慢地装入有刻度的量筒中,并将量筒水平匀速晃动使微胶囊粉末自然下沉,测定体积,并计算单位体积微胶囊的质量即微胶囊的堆积密度。每一样品进行3次平行实验。
1.3.4 紫苏油微胶囊的流动性测定
准确称取5 g微胶囊样品倒入漏斗,使微胶囊通过漏斗自然下落,在水平圆板上堆积。每一样品进行3次平行试验。测量粉堆高度H及粉堆覆盖半径,按公式求出休止角,休止角θ越大散落性越好,休止角θ越小散落性越差。休止角公式如下:
θ=tan-1(H/R)
式中:H为粉堆高度/mm;R为粉堆覆盖半径/mm。
1.3.5 紫苏油微胶囊溶解度的测定
已知微胶囊的含水量B,准确称量微胶囊质量为W。倒入50 mL的烧杯中,量取38 mL蒸馏水,水温控制在25~30 ℃,多次溶解后转入50 mL离心管中。将上述离心管置于离心机中,以4 000 r/min离心10 min,倾去上清液,重复此步骤至沉淀物不再溶解为止,用少量水将沉淀移入已知质量的蒸发皿中,至于105 ℃烘箱中烘至恒重。
溶解度={1-(W2-W1)/[(1-B)×W]}×100%
式中:W为样品质量/g;W1为称量皿质量/g;W2为称量皿和不溶物质量/g;B为样品含水量。
1.3.6 紫苏油微胶囊包埋率测定
包埋率是指微胶囊所包埋芯材的量与总芯材的量之比,是判断微胶囊包埋芯材效果的关键指标[16]。
包埋率=(样品总油含量-表面油含量)/样品总油含量×100%
1.3.6.1 表面含油量测定
精确称取0.5 g微胶囊样品(m0)于干燥的装有滤纸的漏斗中,将100 mL石油醚分5次加入过滤至干燥的旋蒸瓶(m1)。在50 ℃水浴中用旋转蒸发仪蒸干溶剂,65 ℃烘箱中烘干至恒重,冷却称重(m2)。每一样品进行3次平行试验。微胶囊表面油含量的计算公式为:
表面油含量=(m2-m1)/m0×100%
1.3.6.2 样品总油含量测定
索氏抽提法[17]:按照GB/T 5009.6—2016《食品中脂肪的测定》的第一法执行,并进行了适当的修改。
称取0.5 g微胶囊样品(m0),充分研磨10 min,全部移入滤纸筒内(m1)。以无水乙醚作溶剂开始进行索氏抽提。抽提结束以后,将滤纸包取出,65 ℃烘箱中烘干至恒重,称质量(m2)。每一样品进行3次平行试验。微胶囊样品总油含量的计算公式为:
样品总油含量=(m2-m1)/m0×100%
1.3.7 紫苏油微胶囊的抗潮性实验
准确称取微胶囊产品10 g左右,放在25 ℃,80%相对湿度的恒温恒湿箱中,测定一定时间内微胶囊产品增加的质量,计算其增重率。每一样品进行3次平行试验。
1.3.8 紫苏油微胶囊的囊壁耐热性实验
准确称取微胶囊产品1 g,用锡纸包裹,分别置于50、100、150、200、250、300、350 ℃的恒温沙浴锅中保持1 h,测定其包埋率,用来检验微胶囊囊壁的耐热性。
1.3.9 紫苏油微胶囊的模拟消化吸收实验1.3.9.1 模拟胃液的制备[18]
取2.0 g NaCl与7 mL 36%的HCl添加在900 mL的去离子水中,随后再放入3.2 g含量的胃蛋白酶,用0.1 mol/L的HCl调节溶液的pH为1.2,定容到1 000 mL,并放在4 ℃下存储。溶液必须保证现用现配,防止酶发生失活的情况,影响最终实验效果。
1.3.9.2 模拟肠液的制备[18]
取800 mL的去离子水,然后把6.8 g K2HPO4溶于其中,再取77 mL的0.2 mol/L的NaOH与10 g胰蛋白酶,保持在4 ℃环境温度下搅拌,随后搁置一夜,用0.1 mol/L NaOH调节溶液的pH为6.8,定容到1 000 mL,并放在4 ℃下存储。溶液必须保证现用现配,防止酶发生失活的情况,影响最终实验效果。
1.3.9.3 模拟消化吸收实验[19]
取1 g紫苏油微胶囊加到100 mL模拟胃液中,使用磁力搅拌器在(37±0.5) ℃、100 r/min的条件下搅拌,每20 min过滤取出微胶囊进行称重,2 h后,再过滤加到100 mL模拟肠液中,使用磁力搅拌器在(37±0.5)℃、100 r/min的条件下搅拌,每30 min过滤取出微胶囊进行称重。每一样品进行3次平行试验。
1.3.10 紫苏油微胶囊释放率的测定
释放油脂的提取方法[20]:间隔0.5 h取混合均匀消化液,将酶灭活,将消化过的样品溶液转入分液漏斗中,加入25 mL正己烷,混合萃取,重复3次后,合并有机相,55 ℃旋蒸除去正己烷,用99.9%的氮气除去残留的溶剂,得到消化过程中释放的油脂含量,释放率(Q)根据以下公式计算:
释放率=消化后油脂释放总量/微胶囊总油含量×100%
1.3.11 微胶囊化前后紫苏油的脂肪酸组成分析1.3.11.1 紫苏油微胶囊破壁提油
准确称取5 g紫苏油微胶囊样品于干燥的研钵中,充分研磨20 min后,转移到干燥的锥形瓶中,加入200 mL石油醚使溶解,超声震荡1 h,过滤。用60 mL石油醚分3次加入洗涤滤渣,每次均充分震荡,过滤并合并滤液于干燥的旋蒸瓶中。在40 ℃水浴中用旋转蒸发仪蒸干绝大部分溶剂,再使用氮吹仪将残余溶剂完全蒸发。
1.3.11.2 紫苏油甲酯化方法
称取30 mg样品,加入1.5 mL 0.5 mol/L氢氧化钠-甲醇溶液,充分混合后在沸水中反应3 min,冷却;再加入2 mL体积分数14%的三氟化硼-甲醇溶液,充分混合后继续在沸水中反应2 min,冷却;再加入1 mL饱和氯化钠和2 mL异丙醇,充分混合后,超速离心,3 000 r/min反应2 min;取上层脂肪酸甲酯,经过无水硫酸钠干燥后用于气相(GC)分析。
1.3.11.3 气相色谱分析条件
检测器:氢离子火焰检测器(FID),260 ℃;色谱柱:HP-5毛细管柱(TR-TRACE-FAME,25 m×0.32 mm×0.50 μm);进样口温度:250 ℃:进样量:1 μL:分流比:30∶1;程序升温:140 ℃,保持5 min,以 3 ℃/min 升至 240 ℃,保持65 min;载气:高纯氦气,流速 34 mL/min。
1.3.12 数据统计和分析
所有实验做3次平行,使用Origin 2018绘制图表。
2 结果与分析
2.1 紫苏油微胶囊基本理化指标
由表1可知,在以明胶、海藻酸钠为壁材,通过复凝聚法制备得到的紫苏油微胶囊产品的含水量较低,为3.91%。这说明产品在冷冻干燥过程中水分充分蒸发,达到了所需的干燥状态,有利于产品的贮藏。休止角是衡量粉末产品流动性的重要指标,一般来说,当休止角≤30°时,产品的流动性很好;休止角为30°~45°时,产品流动性良好;休止角为45°~60°时,产品流动性一般;而休止角≥60°时则流动性差[21]。本实验所得的紫苏油微胶囊休止角为44.47°,这是因为冷冻干燥的微胶囊产品表面存在褶皱,并不光滑,因此产品的流动性勉强接近良好。微胶囊的溶解度很低,几乎不溶于水,这是由于该微胶囊外壁选用复合材料,因此在单纯的水中经过凝聚固化的复合壁材很难溶解,起到了对芯材的保护作用。
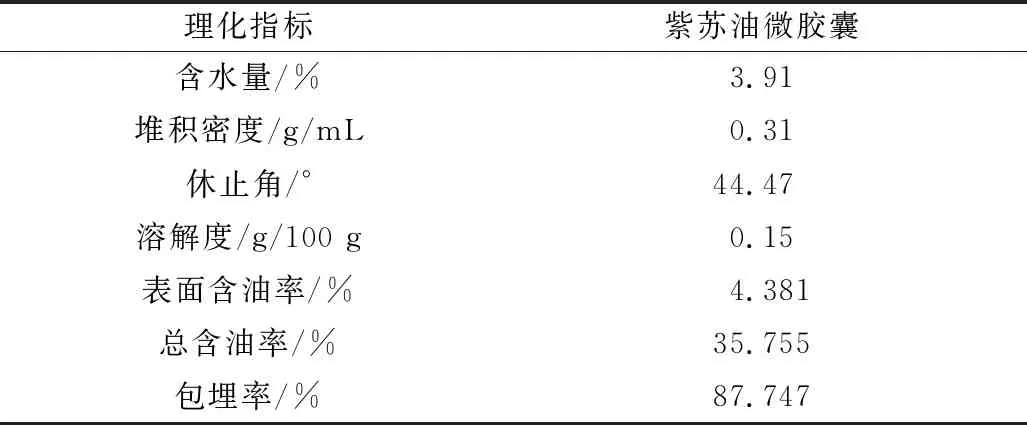
表1 紫苏油微胶囊基本理化指标
2.2 紫苏油微胶囊抗潮性分析
由图1可知,紫苏油微胶囊在初始阶段水分增重较快,这是因为壁材物质明胶和海藻酸钠吸水性良好,随着其吸水能力逐渐饱和,微胶囊的增重也逐渐缓慢,最终趋向恒定。食品中含水量的大小与其货架期密切相关。对于油脂微胶囊产品来说,壁材吸水后其韧性减弱甚至发生损坏,失去对芯材的保护作用;芯材易于跟壁材吸收的水分接触,容易发生氧化酸败,这样会缩短产品的货架期,影响微胶囊产品的质量。因此,微胶囊产品应尽量置于干燥的环境中保存。
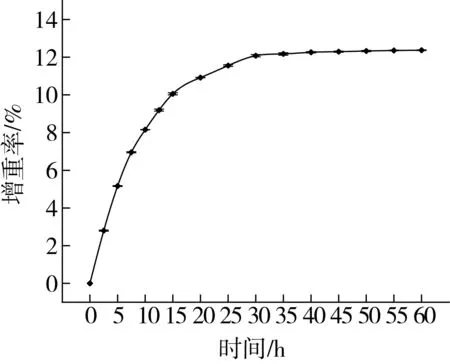
图1 紫苏油微胶囊抗潮性曲线
2.3 紫苏油微胶囊的囊壁耐热性分析
由图2可知,在刚开始阶段,微胶囊在50 ℃下,包埋率仅下降了1.3%,几乎没有损失;随着温度的升高,在50~150 ℃期间,包埋率开始缓慢下降,这可能是壁材受热发生微小变化或者部分分解,或者表面水分蒸发等,使囊壁结构发生改变,导致部分芯材被释放出来从而降低了包埋率。在温度为150~250 ℃期间,曲线迅速下降,包埋率降低了60%,这可能是明胶和海藻酸钠间由于静电力作用形成的共价键遭到破坏,发生了热分解,被包埋的紫苏油随着壁材的破坏而大量释放。当温度超过250 ℃后,曲线开始趋于平缓,这是由于微胶囊结果破损严重,芯材已经损失殆尽,包埋率变化波动变小。

图2 不同温度下紫苏油微胶囊包埋率的变化
因此,在紫苏油微胶囊的储存过程,短暂的夏季高温对其质量影响不大,但应尽量置于阴凉处;而将紫苏油微胶囊作为食品原辅料进行加工的时候则需注意,温度应该控制200 ℃以下,且尽可能缩短加工时间,以减少高温对紫苏油微胶囊带来的不利影响。
2.4 紫苏油微胶囊在模拟消化吸收过程中的释放性能分析
2.4.1 紫苏油微胶囊在模拟消化吸收过程中的累积释放率分析
0~2 h为紫苏油微胶囊进行体外人工模拟胃液消化阶段,由图3可知,体外人工模拟胃液消化后,微胶囊释放率为13.25%。表明以明胶—海藻酸钠为壁材制得的微胶囊,其囊壁外壳具备耐胃液消化分解的能力。2~5 h为紫苏油微胶囊进行体外人工模拟肠液消化阶段,由显著升高的曲线可以看出,紫苏油微胶囊在肠液中,囊壁迅速崩解,将包裹的芯材释放在肠道中。体外人工模拟肠液消化后,微胶囊释放率为33.02%。表明该微胶囊能够保护PUFA和其他亲脂性成分顺利完整的到达小肠再释放出来进行消化吸收。因此,紫苏油微胶囊在消化吸收过程中具有较好的释放率。
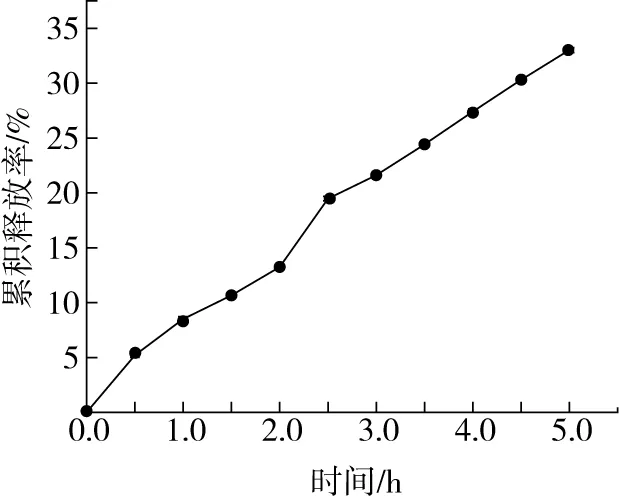
图3 紫苏油微胶囊在模拟消化吸收过程中的累积释放率
2.4.2 紫苏油微胶囊在模拟消化吸收过程中释放性能分析
将紫苏油微胶囊在模拟胃液环境和肠液环境中的释放数据进行零级释放模型、Higuchi 模型和Ritger-Peppas模型拟合[22],拟合曲线如图4、图5,拟合结果如表2。

图4 紫苏油微胶囊在模拟胃液中的释放拟合曲线
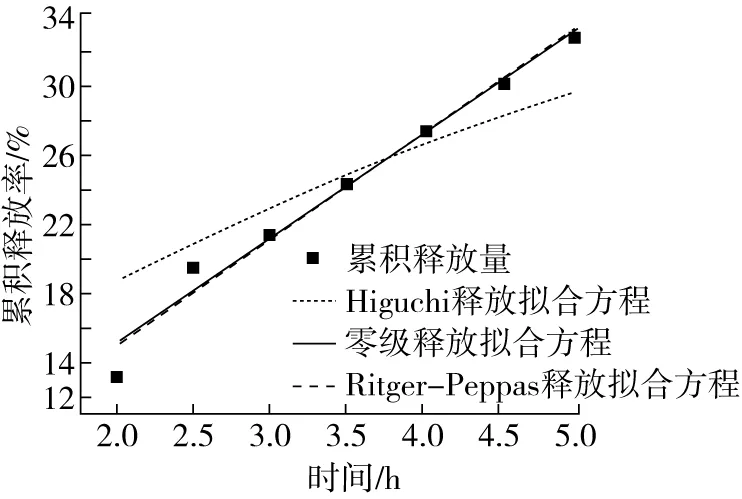
图5 紫苏油微胶囊在模拟肠液中的释放拟合曲线
由图4、图5可知,紫苏油微胶囊在胃液中2 h时,累积释放率为 13.25%;紫苏油微胶囊在肠液中5 h时,累积释放率达到33.02%。可知紫苏油微胶囊在胃液中释放较慢,大部分在肠道中释放。

表2 紫苏油微胶囊在模拟消化吸收过程中的释放模型拟合结果
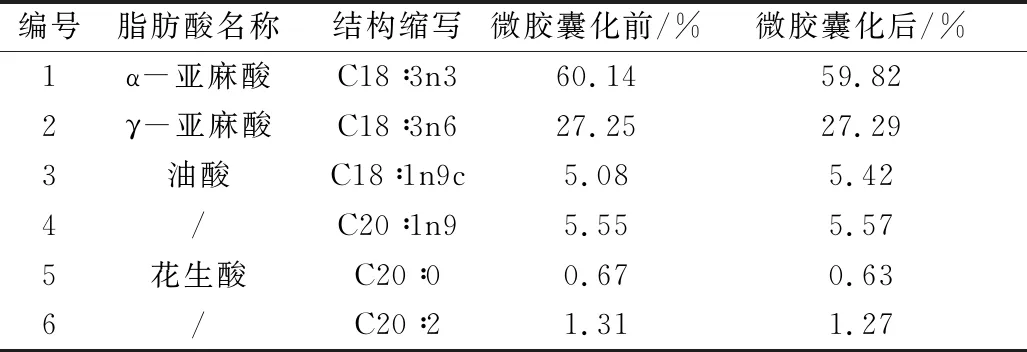
通过比较表2中模拟胃液环境中的拟合系数R2可知,Ritger-Peppas模型>零级模型>Higuchi模型,因此紫苏油在胃液中的释放最符合Ritger-Peppas模型,拟合系数R2为0.998 5。Ritger-Peppas方程的释放参数n=0.66,当0.45 由表3可知,微胶囊化之前,紫苏油的主要功能性成分α-亚麻酸和γ-亚麻酸质量分数分别为60.14%和27.25%;经过微胶囊化之后,α-亚麻酸和γ-亚麻酸质量分数分别为59.82%和27.29%;其他脂肪酸含量虽然有少量变化,但基本保持不变。由此可见,采用温和的复凝聚法和冷冻干燥制备微胶囊,对紫苏油的脂肪酸营养价值并未造成破坏,对芯材起到了很好的保护作用。 表3 微胶囊化前后紫苏油脂肪酸成分的变化 本研究对紫苏油微胶囊进行理化特性、模拟胃肠道条件下的消化吸收能力分析以及微胶囊化前后紫苏油脂肪酸成分进行分析。结果表明,紫苏油微胶囊含水量较低,易于产品的贮藏;产品的黏度较小,流动性较好;包埋率为87.747%。模拟消化吸收实验结果表明,释放机理符合Ritger-Peppas模型,紫苏油微胶囊具有良好的释放性,并且在模拟消化吸收过程中的释放扩散是一个相对稳定的过程。微胶囊化前后紫苏油的脂肪酸成分分析结果表明,脂肪酸含量基本无变化,营养价值未受到破坏,对芯材起到了很好的保护作用。采用复凝聚法对紫苏油实现良好包埋,微胶囊化后样品达到功能性油脂在小肠内有效消化吸收的目的。2.5 微胶囊化前后对紫苏油脂肪酸成分的影响
3 结论
