代谢工程改造嘌呤合成途径以提高核黄素产量
2022-06-15谷振宇夏苗苗苏媛刘川罗华军张大伟
谷振宇,夏苗苗,苏媛,3,刘川,4,罗华军,张大伟,4*
1(三峡大学 生物与制药学院,湖北 宜昌,443002)2(中国科学院 天津工业生物技术研究所,天津,300308) 3(天津科技大学 生物工程学院,天津, 300457)4(中国科学院大学,北京, 300192)
核黄素,又名维生素B2,是人和哺乳动物生长必不可少的一种营养物质,在生物体内参与多种氧化还原反应,尤其在作为黄素酶类的辅酶参与细胞中传递氢的相关代谢中起到了重要作用[1]。核黄素转化为活性形式:黄素腺嘌呤二核苷酸和黄素单核苷酸可以参与促进生物体内碳水化合物的合成、脂肪酸代谢以及呼吸电子链传递[2]。由于哺乳动物自身不能合成核黄素,因而其在饲料行业、食品医药行业中都着有广泛的应用[3-4]。
鸟苷三磷酸(guanosine triphosphate, GTP)是核黄素生物合成的直接前体[5],它既可以以糖和氨基酸为底料进行从头合成,也可以利用嘌呤碱为前体进行补救合成。嘌呤从头合成途径将戊糖磷酸途径生成的5-磷酸核糖最终转化成GTP和腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)。以合成GTP为例,其主要可以分为嘌呤操纵子催化5-磷酸核糖-1-焦磷酸(5-phosphoribosyl 1-pyrophosphate,PRPP)生成肌苷酸(inosincacid,inosinemonphosphate,IMP),以及IMP催化生成GTP两个阶段。发酵过程中添加GTP可以提高核黄素产量[6-7],证明嘌呤前体的有效供应对核黄素的生产至关重要。并且利用代谢工程策略增强GTP的合成也已有很多报道[8-10]。
本文选用的核黄素高产菌株S1在5 L发酵罐中发酵52 h,核黄素产量可达27 g/L。在此基础上利用基于CRISPR/Cas9的碱基编辑器失活嘌呤操纵子阻遏蛋白基因以及鸟嘌呤核苷酸(guanylate,GMP)还原酶来分别提高GTP合成中2个阶段的通量。另一方面尝试过表达GMP到2,5-二氨基-6-(5-磷酸-D-核糖氨基)嘧啶-4(3H)酮(DARPP)合成途径的基因以增强GTP的合成。
1 材料与方法
1.1 材料
1.1.1 实验菌株与质粒
论文中涉及的菌株见表1,质粒见表2。

表1 本实验所用菌株Table 1 Strains used in this study

表2 本实验所用质粒Table 2 plasmids used in this study
1.1.2 引物
本实验所涉及的基因序列均来自NCBI(https://www.ncbi.nlm.nih.gov/),使用SnapGene设计所用引物,由金唯智生物公司合成(表3)。

表3 本实验所用引物Table 3 Primers used in this study
1.1.3 主要培养基
LB培养基(g/L):酵母提取物5,蛋白胨10,NaCl 10;灭菌前用NaOH溶液调节pH值至7.2,121 ℃灭菌20 min。
SX培养基(g/L):酵母提取物5,蛋白胨10,NaCl 5;灭菌前用NaOH溶液调节pH值至7.2,121 ℃灭菌20 min。
发酵培养基YP(g/L):玉米浆干粉5,蔗糖10、硫酸镁5,酵母抽提物5,K2HPO43,KH2PO42,灭菌前用NaOH溶液调节pH值至7.2,121 ℃灭菌20 min。
补料培养基(g/L):一水葡萄糖720,玉米浆干粉15,KH2PO40.3,灭菌前用NaOH溶液调节pH值至7.2,115 ℃灭菌30 min。
1.1.4 仪器与设备
GET3XG三槽独立梯度基因扩增仪,杭州柏恒科技有限公司;水平电泳仪,北京市六一仪器厂;94-Z水平摇床,宁波新芝生物科技股份有限公司;V-1600可见分光光度仪,上海美谱达仪器有限公司;SBA-40E 生物传感分析仪,山东生物传感器重点实验室。
1.2 实验方法
1.2.1 嘌呤合成途径基因过表达及失活菌株的构建
1.2.1.1 质粒构建
以SG质粒为例,以pHP13-spe-PvegI-mcherry质粒为模板,用引物pHP131、PvegI-2扩增含壮观霉素抗性基因以及PvegI启动子的pHP13质粒骨架,以B.subtilis168基因组为模板,用引物UP-gmk、DN-gmk扩增gmk基因片段,将扩增出来的2个片段用Gibson试剂盒两片段连接,化学转化DH5α后,挑取长出的菌落用CXPHP131、CXPHP132引物进行验证,正确的转化子大小为1 293 bp,验证且测序正确后接入无抗的LB培养基中培养14~17 h提取质粒。
1.2.1.2 嘌呤合成途径基因过表达
向目的菌株中用电转化[11]的方法转入相应基因的过表达质粒,以终质量浓度为150 μg/mL的壮观霉素进行筛选获得正确的转化子后保藏。
1.2.1.3 碱基编辑器质粒的构建及嘌呤合成途径基因失活
碱基编辑器失活guaC和purR的质粒基于实验室中构建的碱基编辑器质粒pHP13-spe-dcas9。质粒以pHP13为骨架包含复制起始位点Puc ori,复制蛋白Pep pTA1060,壮观霉素抗性基因spe。其余部分包括乳糖/IPTG诱导的阻遏蛋白基因lacI,PgraC启动子表达dcas9及脱氨酶(adenosine deaminase,AID),其中dcas9缺失终止密码子,PvegI启动子连接仅缺少N20的向导RNA。在插入N20序列时,首先需要找到相应基因序列前中段合适的NGG序列,NGG序列上游20 bp为N20序列,由于在NGG上游15~20 bp更容易利用碱基编辑器发生编辑,所以在NGG上游15~20 bp寻找使C变为T而产生终止密码子的突变,设计N20以构建质粒。以PDC质粒为例,以pHP13-spe-dcas9质粒为模板,用引物UP-dcas9-guaC、DN-dacs9扩增片段1,用引物UP-dac9、DN-dcas9-guaC扩增片段2,将扩增出来的2个片段用Gibson试剂盒两片段连接,化学转化DH5α后,挑取长出的菌落用CX13 spe2、CXJF2引物进行验证,正确的转化子大小为624 bp,验证且测序正确后接入无抗的LB培养基中培养14~17 h后提取质粒。
向目的菌株中用电转化的方法转入相应基因的碱基编辑器质粒,以终质量浓度为150 μg/mL的壮观霉素进行筛选获得正确的转化子后,进行传代培养使目的位点发生编辑并且使质粒丢失,影印后对质粒丢失的菌进行测序,得到相应密码子突变成终止密码子的菌株进行保藏。
1.2.2 核黄素浓度的测定
取500 μL发酵液样品于1.5 mL EP(eppendorf)管中,用0.1 mol/L NaOH溶液稀释适宜倍数,避光碱溶20 min取出,12 000 r/min离心1 min,取上清液用分光光度计检测444 nm处的吸光度值,以确定核黄素的浓度[12]。
1.2.3 残糖测定
取500 μL发酵液样品于1.5 mL EP管中,12 000 r/min离心1 min,取上清液用无菌水稀释适宜倍数后,用进样针取样,用生物传感分析仪对样品的残糖进行检测。
1.2.4 蔗糖测定
取200 μL发酵液样品于1.5 mL EP管中,加入200 μL配制好的2 mol/L H2SO4溶液,于60 ℃水浴锅中水解30 min,加入4 mol/L NaOH溶液200 μL进行中和,12 000 r/min离心1 min,取上清液用无菌水稀释适宜倍数,用生物传感分析仪对样品水解后的葡萄糖进行检测,蔗糖的浓度为测得的葡萄糖浓度的2倍。
1.2.5 摇瓶补料发酵验证
从-80 ℃冻存管中取3 μL菌液于含有1 mL无菌水的EP管中,10倍稀释后混匀,取3 μL涂布SX平板,37 ℃倒置培养48 h,挑取直径5~6 mm的1个单菌落接种2支固体试管SX含相应抗性的斜面培养基,37 ℃静置培养48 h。用1 mL YP培养基清洗下一支斜面上的所有菌苔于2 mL EP管中,混匀,取300 μL菌液分别接种于含有80 mL YP培养基的500 mL瓶底有挡板的三角瓶中,37 ℃,180 r/min振荡培养41 h。培养过程中分别在11和23 h加入补料培养基进行补料发酵。
2 结果与分析
2.1 嘌呤合成途径基因失活及补料发酵验证
根据文献报道,purR基因编码嘌呤合成途径的阻遏蛋白PurR,敲除purR基因可使嘌呤合成途径基因转录水平大幅提升[13-14]:guaC基因为嘌呤合成途径GMP还原酶编码基因,敲除guaC基因[15]可阻断GMP到IMP的合成,从而使GMP得到一定的积累,间接提高核黄素的产量。因此对这2个基因用CRISPER/Cas9介导的基因组碱基编辑器技术进行失活[16-17],分别得到菌株SDR和SDC,并对这2个菌株进行补料发酵验证。
由图1-b可知,嘌呤合成途径的阻遏蛋白purR碱基编辑器失活后核黄素产量从2.81降至2.68 g/L,证明高产菌S1中嘌呤合成途径由PRPP到IMP部分基因转录水平满足核黄素的合成,不是该途径中的限速步骤;而嘌呤合成途径的回补途径基因guaC碱基编辑器失活后,阻断了GMP到IMP的合成,核黄素产量由2.81 g/L提升至3.04 g/L,产量提升8%。41 h时S1菌以及SDR菌葡萄糖有少量剩余,SDC菌葡萄糖剩余2.67 g/L(图1-c),17 h时摇瓶内蔗糖已耗尽。SDC菌株核黄素转化率由S1菌株1 g糖(0.392 g蔗糖、0.608 g葡萄糖)产0.027 5 g核黄素提高至1 g糖(0.399 g蔗糖、0.601 g葡萄糖)产0.030 3 g核黄素,转化率提升10.2%。

a-最大OD600测定值;b-发酵结束核黄素终产量;c-补糖前葡萄糖测定图1 失活基因摇瓶补料发酵Fig.1 Feeding fermentation of inactivated gene in shake flask
2.2 嘌呤合成途径基因过表达
以上实验表明guaC基因失活使产量有所增加,推测GMP的供给并没有达到饱和。基于此推论,尝试将gmk和ndk基因进行过表达(gmk编码鸟苷酸激酶,催化GMP生成GDP,ndk编码核苷二磷酸激酶,催化GDP生成GTP),期望通过增加GMP的消耗拉动IMP向GMP的转化过程。因此对这2个基因以pHP13为质粒骨架,PvegI为启动子构建质粒PG和PN,电转化进S1菌株,分别得到菌株SG和SN,并且用携带不含启动子及目的基因的空载质粒S1-空菌株作为对照,进行补料发酵验证。
由图2-a可知,过表达gmk及ndk后对菌株生长无明显影响。图2-b为核黄素终产量,与对照菌株S1-空相比,SG菌株及SN菌株产量无明显提高,41 h时葡萄糖无剩余(图2-c),17 h时摇瓶内蔗糖已耗尽。这说明单独表达这2个基因并没有拉动GMP的合成,而GTP进入核黄素合成途径可能是更关键的反应。

a-最大OD600测定值;b-发酵结束核黄素终产量;c-补糖前葡萄糖测定图2 gmk、ndk基因过表达摇瓶补料发酵Fig.2 Feeding fermentation of overexpression of gmk and ndk genes in shake flask
2.3 ribA基因异源引入及过表达
根据文献报道以及BRENDA数据库(https://www.brenda-enzymes.info/index.php)查询得到枯草自身RibA双功能酶(EC 3.5.4.25及EC 4.1.99.12),但该酶无法将2个酶的功能拆分,为了将GTP环水解酶II单独过表达,查找到希瓦氏菌来源的ribA(EC 4.1.99.12)。以pHP13为质粒骨架,以PvegI及Pr为启动子分别对希瓦氏菌ribA基因进行过表达,构建质粒PSA和PRA,电转化进S1菌株,分别得到菌株SSA和SRA,用含空载质粒的S1-空菌株作为对照,进行补料发酵验证。
图3-a可知,S1-空菌株最大OD600为32.2,SSA及SRA菌株最大OD600有不同程度的下降,分别为30.2和23.3。表明希瓦氏菌ribA的表达对菌的生长产生一定的影响,可能与GTP在GTP环水解酶II催化下产生的DARPP具有一定的细胞毒性有关。而Pr启动子表达强度强于PvegI启动子,对菌的生长抑制也表现的较为明显,导致核黄素产量下降。图3-b为发酵结束时核黄素终产量,SSA菌产量提升至3.33 g/L,并且发酵结束摇瓶中仍有4.6 g/L的葡萄糖剩余。而SRA菌产量则明显下降至2.69 g/L,耗糖进一步减少,发酵结束摇瓶中葡萄糖剩余18.4 g/L(图3-c),17 h时摇瓶内蔗糖已耗尽。SSA菌株核黄素转化率为1 g糖(0.407 g蔗糖、0.593 g葡萄糖)产0.033 8 g核黄素,转化率较S1提升22.9%。
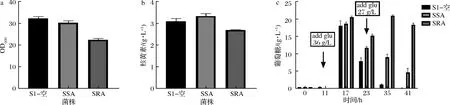
a-最大OD600测定值;b-发酵结束核黄素终产量;c-补糖前葡萄糖测定图3 异源引入基因摇瓶补料发酵Fig.3 Feeding fermentation of heterologous introduction of genes in shake flask
2.4 guaC基因失活及ribA基因过表达组合验证
由以上实验可知,失活guaC基因可以提高GMP的供给,PvegI启动子过表达希瓦氏菌来源的ribA基因可以加快GTP的利用,从而拉动嘌呤途径产物的合成使核黄素产量增加,并且认为这2种效果是可以叠加的。于是在失活guaC基因的SDC菌中分别转入PSA质粒以及PRA质粒,得到菌株SDCA和SDCR,并将pHP13-spe空载质粒转入SDC菌中得到菌株SDCS,用SDCS菌株作为对照,进行补料发酵验证。
图4-a表明,摇瓶补料发酵时Pr启动子表达希瓦氏菌来源ribA在SDC菌中仍会抑制生长。SDCA菌株产量进一步提升,达到3.43 g/L,相较于SDCS菌产量提高9.2%,相较于初始菌株S1产量提升21.7%(图4-b);并且SDCA菌耗糖也有所减少,发酵结束时摇瓶中剩余葡萄糖3.87 g/L。SDCS菌发酵结束时没有葡萄糖剩余(图4-c),SDCA菌株转化率为1 g糖(0.404 g蔗糖、0.596 g葡萄糖)产0.034 6 g核黄素,较S1菌株提升25.8%,具有一定的工业生产应用价值。

a-最大OD600测定值;b-发酵结束核黄素终产量;c-补糖前葡萄糖测定图4 组合基因摇瓶补料发酵Fig.4 Feeding fermentation of combined gene in shake flask
3 结论与讨论
S1是1株经过诱变及筛选所得的枯草芽孢杆菌核黄素高产菌株,但核黄素合成必须的鸟嘌呤合成途径并未发生突变。为了使S1菌株核黄素合成能力进一步提升,主要对嘌呤合成途径的相关基因进行失活以及质粒过表达,结果如下:
(1)嘌呤合成途径支路基因失活:运用CRISPER/dCas9介导的基因组碱基编辑器技术失活guaC基因(编码GMP还原酶),阻断了GMP到IMP的合成,核黄素产量由2.81提升至3.04 g/L,提升8.2%,转化率提升10.2%。
(2)嘌呤合成途径下游基因过表达:通过质粒将GMP下游合成基因gmk(鸟苷酸激酶)和ndk(核苷二磷酸激酶)进行过表达,核黄素产量没有明显提升,由此确定了这两步反应不是核黄素合成的瓶颈。
(3)希瓦氏菌ribA基因引入及过表达:枯草芽孢杆菌中ribA基因编码的蛋白为一种双功能酶,希瓦氏菌中ribA(EC 3.5.4.25)和ribB(EC 4.1.99.12)2种酶可以拆分单独存在,故单独引入了希瓦氏菌ribA基因(EC 3.5.4.25),过表达GTP环水解酶II部分,将GTP更多的转化为DARPP,增强嘌呤前体的利用。在摇瓶补料发酵中核黄素产量由3.09提升至3.33 g/L,并且耗糖有所减少,发酵结束时葡萄糖剩余4.6 g/L,核黄素转化率较S1提升22.9%
(4)将guaC基因失活与希瓦氏菌ribA基因过表达进行组合,核黄素产量进一步提升至3.43 g/L,较初始菌株S1产量提升21.7%,并且耗糖有少量减少,核黄素转化率提升25.8%,具有工业生产价值。
