Ru 掺杂对Pt(111)面CO 覆盖度影响的理论研究
2022-06-08丁浩然姚陈忠
丁浩然,姚陈忠
(运城学院 应用化学系,山西 运城 044000)
引言
能源供需不平衡、化石燃料面临耗竭和环境污染等问题是当今人类社会面临的主要挑战。因此,寻求更高效、更安全、更清洁甚至“ 零排放” 的新能源技术,是实现人类可持续发展的关键[1-2]。直接甲醇燃料电池(DMFC)是一种将甲醇中储存的化学能直接转化为电能的洁净环保器件,因其不受卡诺循环的限制,兼具高转换效率与低废物排放等优点,得到研究者越来越多的关注[1-3]。
DMFC 的关键组成部分之一是阳极甲醇氧化反应的电催化剂。甲醇氧化反应由于其六电子转移过程而在动力学上速度缓慢[3-4]。Pt 基催化剂由于其优异的甲醇电催化性能,是目前研究的热点。研究发现,Pt 的催化活性与颗粒大小、形貌、结构和表面状态密切相关,特别是高的CO 覆盖度将会导致催化剂快速失活[5]。为了解决这一问题,一种策略是开发新型Pt 基双金属材料,其中Pt 作为甲醇氧化活性中心,另一金属成分作为CO 去除剂。利用这种协同效应,该催化剂同时对甲醇氧化反应表现出良好的活性和优良的耐久性。其中,Pt-Ru 双金属纳米材料被认为是迄今为止性能最好的抗CO 中毒催化剂[6-7]。
基于此,文章首先通过理论计算,系统研究了Ru 掺杂对Pt 电子结构的影响;随后计算掺杂前后CO 吸附能和CO 活化转化的差异;最终通过理论计算解释Ru 掺杂能够降低Pt 催化剂表面CO 覆盖度的原因。
1 计算方法和模型
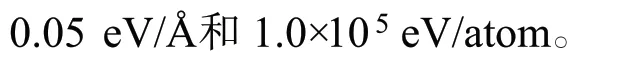
因为X 射线衍射仪表征结果显示Pt(111)面是金属Pt 的主要暴露晶面[12],所以计算使用Pt(111)面。Pt(111)面采用的4 层周期性模型,真空层厚度为1.5 nm。计算时,固定Pt(111)面的最下两层,其余原子和吸附分子可以弛豫。我们考虑了不同吸附位的吸附(图1):顶位(top),桥位(bridge),三重穴位hcp 和fcc,在文中只比较最稳定的吸附位。
图1 Pt(111)面的俯视(a)和主视图(b)
吸附能的计算公式是:Eads= E(mol)+ E(slab)-E(mol/slab),其中E(mol),E(slab)和E(mol/slab)分别为自由分子的能量、底物的能量和吸附物吸附在底物上时系统的总能量。因此,吸附能越大说明吸附物和底物之间的相互作用力就越大。
2 结果与讨论
首先笔者研究Ru 掺杂对Pt(111)面电荷结构的影响。因为一个Ru 掺杂在最上层,所以我们只讨论第一层原子的电子结构。掺杂前,bader 电荷显示最上层每个Pt 的电子为-0.06 e,这说明在Pt(111)表面有部分电子的聚集。掺杂后,Ru 的bader 电荷为0.46 e,Pt 的电荷约为-0.11 e。bader电荷结果显示,掺杂后Ru 的电子向Pt 转移。这是因为Ru 和Pt 的电负性为1.54 和1.72,后者的电负性大于前者,所以Ru 的电子向Pt 转移。
图2 是Pt(111)和Ru-Pt(111)面第一层原子的分波态密度(PDOS)。比较图2(a)和图2(b)可以看到,由于掺杂量较低,Pt 的PDOS 图没有明显变化。然而,Pt(111)面的d 带中心远离费米能级。这是因为掺杂Ru 后,表面Pt 原子的bader 电荷增加,因此其d 带中心从-2.08 eV 迁移到-2.13 eV。因为Ru 原子的d 带中心位于-1.28 eV(图2c),所以造成掺杂后第一层所有原子的d 带中心又靠近费米能级(-2.13 vs. -2.04 eV)。总体来说,Ru 原子掺杂可以使表面原子的d 带中心靠近费米能级,从而可能对提高催化剂的稳定性有利[13]。

图2 Pt(111)和Ru-Pt(111)面第一层原子的分波态密度

首先,我们考虑掺杂前后CO 吸附能的变化。CO 以C 端吸附在Pt(111)面,其最稳定吸附位是fcc 位 (图 3a)。此时,Pt-C 键键长为0.211 nm,对应的吸附能为1.82 eV。考虑熵的贡献,300 K时CO 的吸附能为1.20 eV。高的吸附能说明CO在常温时很难从Pt 表面脱附,从而导致DMFC电催化剂极易中毒。掺杂Ru 后,CO 仍以C 端吸附,此时Pt-C 和Ru-C 键长分别为0.216 nm 和0.206 nm (图 3d),吸附能等于1.81 eV。Bader 电荷结果显示,掺杂前Pt(111)表面向CO 转移0.39 e;掺杂后表面向CO 转移0.34 e,这说明掺杂抑制了表面电荷的转移。CO 吸附时,尽管掺杂后Ru-C 键长小于Pt-C 键长,但是Pt-C 键长增加,这可能是CO 吸附能没有变化所致。因此,Ru 的掺杂不能改变CO 在Pt 电极表面的吸附能。

图3 Pt(111)和Ru-Pt(111)面吸附CO、COOH 和CO2 构型图
COOH 的最稳定吸附位是桥位,O 和C 原子分别吸附于相邻的Pt 原子顶位,OH 指向Pt(111)面 (图 3b)。Pt-C 和Pt-O 键长分别为0.197 nm 和0.224 nm,对应的吸附能为2.88 eV。当COOH吸附在Ru-Pt(111)面时,其吸附构型没有发生变化,但是O 倾向于吸附在Ru 位。此时,Pt-C 和Ru-O 键长分别为0.197 nm 和0.214 nm (图 3e),吸附能为3.25 eV。掺杂后,Pt-C 键长没有变化,但是Ru-O 键长缩短,这可能是吸附稳定性增加的一个原因。Bader 电荷结果显示,Pt(111)面吸附COOH 时,Pt 向COOH 转移0.08 e;当COOH吸附在Ru-Pt(111)面时,其向COOH 转移0.12 eV。因此,键长的减少和电荷转移数量的增加造成COOH 在Ru-Pt(111)面的吸附增加。
CO2在Pt(111)和Ru-Pt(111)面上吸附时(图3c 和图3f),其分子远离表面,属于物理吸附,吸附能分别只有0.08 eV 和0.02 eV。Bader 电荷也显示CO2吸附前后,二者之间没有的电荷转移,这与吸附能计算结果一致。
对于基元反应来说,反应热的定义为

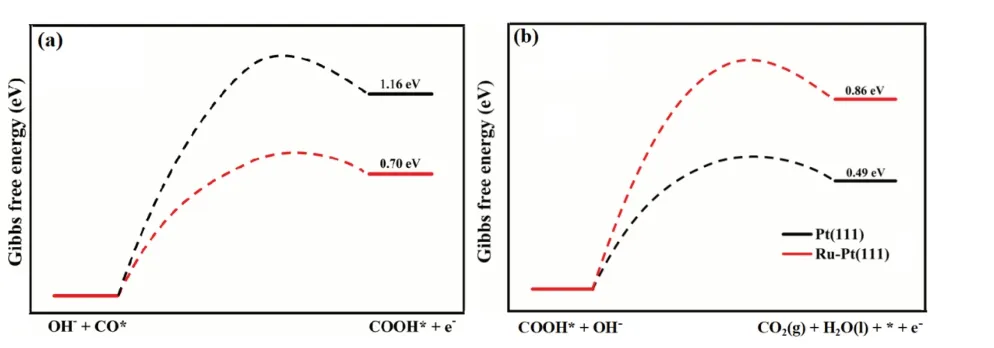
图4 Pt(111) 和Ru- Pt(111)面OH- + CO* → COOH* + e-和COOH* + OH- → CO2 (g) + H2O (l) + * + e-的反应热(a 图缺标注Pt(111)和Ru-Pt(111)


3 结论
基于DFT 计算,文章详细的讨论了Ru 掺杂对CO 在Pt(111)面覆盖度的影响。我们从两个方面考虑了降低CO 覆盖度的方法:对CO 吸附能力和转化活化能的影响。研究结果如下:
(1)Ru 掺杂后,Ru 核外电子向Pt 转移,使Pt 核外电子富集。同时,Ru 掺杂使电极表面的d 带中心向费米中心偏移,从而提高其催化活性。
(2)Ru 掺杂不会影响CO 和CO2的吸附能,但是能够提高COOH 的吸附能。
(3)基于BEP 理论,Ru 掺杂不仅改变了反应的速控步骤,同时有利于降低CO 的覆盖度。
