对氯苯腈的双色共振双光子电离和质量分辨阈值电离光谱*
2022-06-04赵岩李娜党思远杨国全李昌勇3
赵岩 李娜 党思远 杨国全 李昌勇3)‡
1) (晋中学院,物理与电子工程系,晋中 030619)
2) (山西大学激光光谱研究所,量子光学与光量子器件国家重点实验室,太原 030006)
3) (山西大学,极端光学协同创新中心,太原 030006)
本文利用双色共振双光子电离和质量分辨阈值电离光谱技术,研究了对氯苯腈分子第一电子激发态S1 和离子基态D0 的振动特征,确定了对氯苯腈分子S1 ← S0 电子跃迁的激发能为35818 ± 2 cm–1,精确的绝热电离能为76846 ± 5 cm–1.对氯苯腈分子35Cl 和37Cl 两种同位素有相同的激发能和电离能以及相似的振动特征.采用高精度密度泛函方法,计算了对氯苯腈分子在中性基态S0、第一电子激发态S1、离子基态D0 的结构参数和振动频率,分析了电子激发和电离过程中对氯苯腈分子结构和振动频率的变化,并对激发态和离子基态的振动光谱进行了归属,振动光谱上的活性振动大多数是苯环平面内的弯曲振动.通过比较对氯苯酚、对氯苯胺、对氯苯甲醚、对氯苯腈与苯酚、苯胺、苯甲醚、苯腈分子的跃迁能,分析了取代基Cl 原子与苯环之间的相互作用及其对分子跃迁能的影响.
1 引言
苯及其衍生物分子一直是激光光谱实验和理论研究的重点,用于分析分子的光物理和光化学性质,例如激发态分子内电荷转移[1,2]、光解离动力学[3,4]、氢键相互作用[5,6]、光异构[7,8]等.近年来,为了深入研究苯及其衍生物分子的物理化学性质,采用激光诱导荧光、共振增强多光子电离、时间分辨瞬态吸收、时间分辨光电子成像、质量分辨阈值电离光谱技术获得了大量苯衍生物分子激发态和离子态的光谱[9−13].对氯苯腈(p-chlorobenzonitrile)是苯腈分子上与氰基(CN)对位的氢原子被氯原子取代形成的苯衍生物分子.Findley 等[14]通过光电子能谱技术,测量了对氯苯腈分子的光电离曲线,由于光电子能谱的分辨率较差,获得的分子电离能不精确,并且无法观察到离子态的振动光谱.Masao 等[15]利用微波光谱技术测量了对氯苯腈分子的转动常数.2014 年,Rocha 等[16]研究了邻氯苯腈、间氯苯腈、对氯苯腈分子多聚体中分子间的相互作用.2015 年,Trivedi 等[17]测量了对氯苯腈分子的红外光谱和紫外可见光谱,分析了对氯苯腈分子的物理性质和热学性质,评估对氯苯腈分子用于生物场治疗的可能性.但是,对氯苯腈分子在第一电子激发态S1和离子基态D0的振动光谱还尚未有文献报道.
在本文中,采用共振双光子电离和质量分辨阈值电离光谱技术,通过高分辨的飞行时间质谱,分别测量了对氯苯腈35Cl和37Cl 同位素分子的激发态和离子基态光谱.实验确定了对氯苯腈分子S1←S0跃迁的激发能和精确的绝热电离能,并对振动光谱进行了振动模式归属,讨论了电子跃迁引起的分子结构变化.
2 实验方法与理论计算
实验装置主要由真空系统、激光系统和信号采集系统组成[11,18,19].对氯苯腈样品由Sigma-Aldrich公司生产,纯度为99%,常温下对氯苯腈分子是白色晶状固体,将样品装入样品池中,加热到约140 ℃以产生足够高的蒸汽压.使用2 bar (1 bar=105Pa)的氩气作为载气,携带样品分子经过一个脉冲阀(General valve,直径0.5 mm)进入真空系统的束源室,绝热膨胀产生超声分子束.超声分子束经过直径为1 mm 的撇取器进入电离室,与激光束相互作用发生电离.在实验过程中,脉冲阀的打开时间为150 µs,重复频率为10 Hz,束源室与电离室的真空度分别为1.2×10–4Pa 和1.5×10–6Pa.
采用双色共振双光子电离(two-color resonance enhanced two-photon ionization,2C-R2PI)光谱技术测量对氯苯腈分子的第一电子激发态光谱,第一束激发光激发对氯苯腈分子跃迁到第一电子激发态,然后激发态的分子吸收第二束电离光电离,通过固定电离光的波长,扫描激发光的波长获得分子激发态的光谱,激发光的波长范围为270—279.5 nm,电离光的波长固定在240 nm.实验中,两束激光都是采用Nd:YAG 激光器(Quantel Qsmart 850)泵浦染料激光器后,经过BBO 晶体二倍频产生的,使用波长计(HighFinesse WS-7)校准两束激光的波长.通过透镜将激发光和电离光松散聚焦到电离室的中心位置,与分子束相互作用,对氯苯腈分子电离后产生的离子信号在离子透镜直流电场的作用下,通过自由飞行区加速到达微通道板MCP 探测器上.
采用质量分辨阈值电离(mass analyzed threshold ionization,MATI)光谱技术测量对氯苯腈分子的离子基态光谱,首先,固定第一束激发光的波长,将对氯苯腈分子激发到第一电子激发态的某一振动能级;然后,扫描第二束电离光的波长,将分子激发到长寿命的高里德堡态,大约200 ns后,采用–0.5 V/cm 的脉冲回拉电场除去直接电离和自电离的离子,约20 µs 后,施加140 V/cm 的脉冲电场使中性的高里德堡态分子电离,产生的阈值离子在高压电场的作用下被MCP 探测器收集,通过多通道计数器(Stanford Research Systems,SR430)记录飞行时间质谱.实验过程中,采用脉冲延迟发生器(Stanford Research Systems,DG645)控制激光的出光时间、脉冲电场的开启时间、信号的采集时间等.
为了分析对氯苯腈分子在第一电子激发态和离子基态的光谱,使用Gaussian 09 软件[20]对对氯苯腈分子进行了结构优化和振动频率计算.对于第一电子激发态S1,采用含时密度泛函理论TDB3LYP/aug-cc-pVDZ 方法,对于分子基态S0和离子基态D0,采用密度泛函理论B3LYP/aug-ccpVDZ 方法.由于振动频率计算忽略了振动非谐性以及基组不完备性,理论振动频率值略高于实验频率值,因此采用合适的校正因子对计算的振动频率进行统一校正[21].
3 实验结果
3.1 对氯苯腈的飞行时间质谱图
由于氯原子存在两种同位素35Cl 和37Cl,对氯苯腈分子也存在两种同位素C7H4N35Cl 和C7H4N37Cl.为了获得高分辨率的飞行时间质谱图,区分对氯苯腈分子中两种同位素的振动光谱,多通道计数器SR430 的时间分辨率设置为5 ns.图1 显示了双色共振双光子电离光谱实验中测量的对氯苯腈分子的飞行时间质谱,质谱图中两个峰分别对应于对氯苯腈分子35Cl 同位素(m/z:137)和37Cl 同位素(m/z:139)的离子信号,表明该飞行时间质谱仪能够对对氯苯腈分子中35Cl 和37Cl 两种同位素的激发态和离子态光谱进行选择性测量.
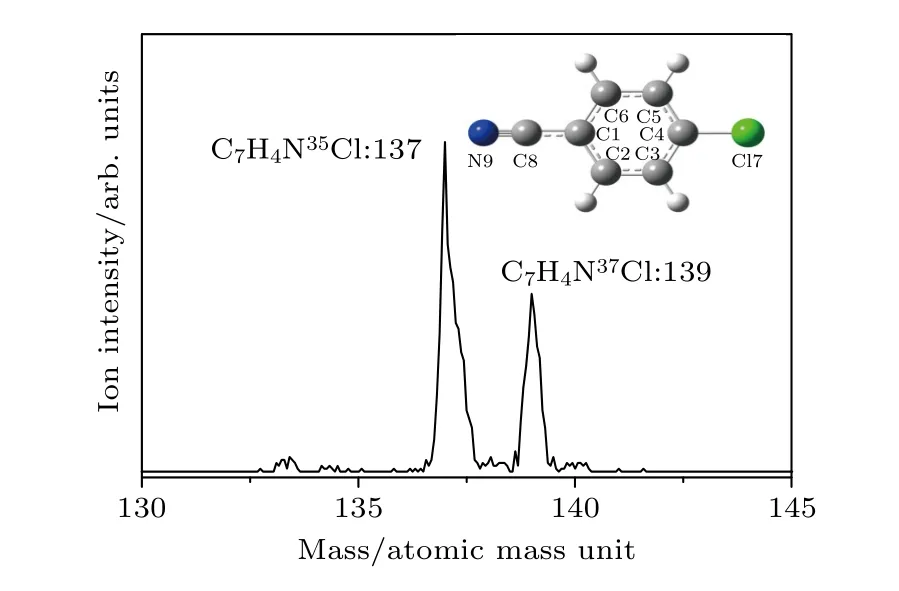
图1 对氯苯腈分子的飞行时间质谱图Fig.1.TOF mass spectrum of p-chlorobenzonitrile.
3.2 对氯苯腈分子第一电子激发态S1 的振动光谱
采用双色共振双光子电离光谱技术,测量了对氯苯腈分子35Cl 和37Cl 两种同位素在S1← S0电子跃迁的带附近的激发态振动光谱,如图2(a)和图2(b)所示.从两个光谱图中可以看出,最强的谱峰分别是对氯苯腈分子35Cl 和37Cl 两种同位素S1← S0电子跃迁的带,相对应的激发能都是35818 ± 2 cm–1,与Maiti 等[22]利用紫外可见吸收光谱技术测量对氯苯腈分子在乙醇溶液中的激发能35640 cm–1接近.通过B3LYP/aug-cc-pVDZ、TD-B3LYP/aug-cc-pVDZ 方法计算了对氯苯腈分子35Cl 和37Cl 两种同位素在中性基态S0和第一电子激发态S1的零点能,计算得到两种同位素激发能的差值仅为0.2 cm–1,小于2 cm–1的实验测量误差,表明实验上对氯苯腈分子两种同位素具有相同的激发能.对于实验测量的光谱精度,根据激光线宽、多普勒加宽和其他实验条件可能带来的误差等因素,并参考了已报道的同类实验研究工作确定为2 cm–1.在其他氯取代的苯衍生物分子中,对氯苯胺、对氯苯酚、氯苯分子[23,24]的35Cl 和37Cl 两种同位素也具有相同的激发能.
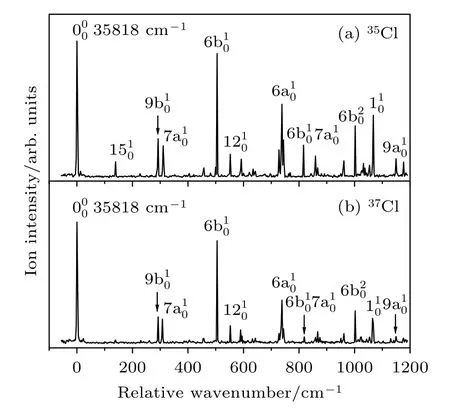
图2 对氯苯腈分子35Cl 同位素(a)和37Cl 同位素(b)的2C-R2PI 光谱,横坐标是相对 带能量的偏移值Fig.2.2C-R2PI spectra of the 35Cl (a) and 37Cl (b) isotopomers of p-chlorobenzonitrile.The spectrum is shifted by 35818 cm-1 (the origin of the S1 ← S0 transition).
对氯苯腈分子有33 个简正振动模式,其中苯环的振动模式30 个,氰基的振动模式3 个.根据Franck-Condon 原理,在这些振动模式中,实验中只能观测到振动重叠积分比较大的电子振动跃迁.对于苯衍生物分子,在S1← S0的电子激发过程中,主要涉及苯环上π 轨道电子的激发[25],引起了与苯环相关的正则振动.依据TD-B3LYP/aug-ccpVDZ 方法计算的振动频率以及之前文献报道的对氯苯胺[23]、对氯苯酚[24]、对氯苯甲醚[26]分子激发态振动光谱的归属,我们指认了对氯苯腈分子35Cl 和37Cl 两种同位素的振动光谱,结果如表1 所列,其中,振动模式的描述和命名采用Wilson标记法[27]和Varsanyi 规则[28].在振动光谱图中,带的右侧是对氯苯腈分子激发态S1的活性振动峰.其中,对氯苯腈35Cl 同位素振动频率为504 cm–1,552 cm–1,739 cm–1,1067 cm–1与对氯苯腈37Cl 同位素振动频率为504 cm–1,552 cm–1,739 cm–1,1066 cm–1的振动峰分别归属为电子振动跃迁,这四个振动模式主要涉及苯环平面内的CCC 弯曲运动.对氯苯腈35Cl 和37Cl 同位素的氰基在苯环平面内振动β(C-CN) 的频率分别为592 cm–1和590 cm–1.频率为139 cm–1,292 cm–1,310 cm–1(35Cl 同位素)和139 cm–1,292 cm–1,309 cm–1(37Cl 同位素)的振动峰分别归属为振动模式15,9b,7a,包含了与取代基有关的C-CN 弯曲、C-Cl 弯曲和C-Cl 伸缩振动.频率为1002 cm–1的振动峰归属为对氯苯腈分子6b 振动模式的二次谐频振动6b2.

表1 对氯苯腈分子35Cl 和37Cl 同位素激发态S1 的振动频率及光谱归属(单位:cm–1)Table 1.The measured vibrational frequencies and assignments for the S1 state of 35Cl and 37Cl isotopomers of p-chlorobenzonitrile (unit:cm–1).
3.3 对氯苯腈分子离子基态D0 的振动光谱
Findley 等[14]通过光电子能谱技术,测量对氯苯腈分子的电离能约为9.50 eV,由于光电子能谱的分辨率较差,获得的电离能不够精确.为了获得对氯苯腈分子精确的绝热电离能,进行了光电离效率实验和质量分辨阈值电离(MATI)实验.通过光电离效率曲线的上升沿位置,确定对氯苯腈分子的大致电离能IE(ionization energy)为(76846 ±10) cm–1.而在MATI 光谱实验中,采用弱回拉脉冲电场除去直接电离的离子,仅探测高里德堡态分子场电离的阈值离子信号,在电离位置处产生一个尖峰,得到一个更精确的电离能.图3 为经过对氯苯腈分子35Cl 和37Cl 同位素中间态S100(35818 cm–1)的MATI 光谱,光谱中最强的谱峰对应于对氯苯腈分子离子态的0+带.考虑实验中回拉脉冲电场的Stark 效应引起分子电离能降低,降低的大小可以通过公式 ΔE=修正(F为电场强度,单位V/cm),得到对氯苯腈35Cl 和37Cl 同位素电离能都为76846 ± 5 cm–1(9.52766 ± 0.0006 eV),与光电离效率实验的测量值一致.通过B3LYP/aug-ccpVDZ 方法计算了对氯苯腈分子35Cl 和37Cl 同位素在离子基态D0的零点能,得到两种同位素电离能的差值仅为0.2 cm–1,小于5 cm–1的实验测量误差,表明实验上对氯苯腈分子两种同位素具有相同的电离能.
MATI 光谱实验也给出了对氯苯腈分子离子基态D0的活性振动信息,如图3 所示,对氯苯腈分子的两种同位素具有相同的振动特征.基于B3LYP/aug-cc-pVDZ 方法计算的振动频率以及对氯苯酚[24]、对氯苯甲醚[29]分子的离子态光谱,我们指认了对氯苯腈分子离子态的振动光谱,表2列出了光谱归属的结果.对氯苯腈分子35Cl 同位素以中间态S100的MATI 光谱中如图3(a)所示,振动频率位于601 cm–1,778 cm–1,1110 cm–1的振动峰分别对应于苯环平面内的弯曲振动模式12,6a,1;振动频率位于1388 cm–1,1608 cm–1的振动峰分别对应于苯环平面内的伸缩振动模式8b,8a;振动频率位于346 cm–1,691 cm–1,1035 cm–1的强振动峰分别归属为与取代基C-Cl 伸缩振动有关的振动7a1以及它的二次谐波振动7a2和三次谐波振动7a3.对氯苯腈分子37Cl 同位素以中间态S100的MATI 光谱如图3(b)所示,振动频率位于343 cm–1,681 cm–1,778 cm–1,1031 cm–1,1116 cm–1和1227 cm–1的振动峰分别对应于振动7a1,7a2,6a1,7a3,11和131.
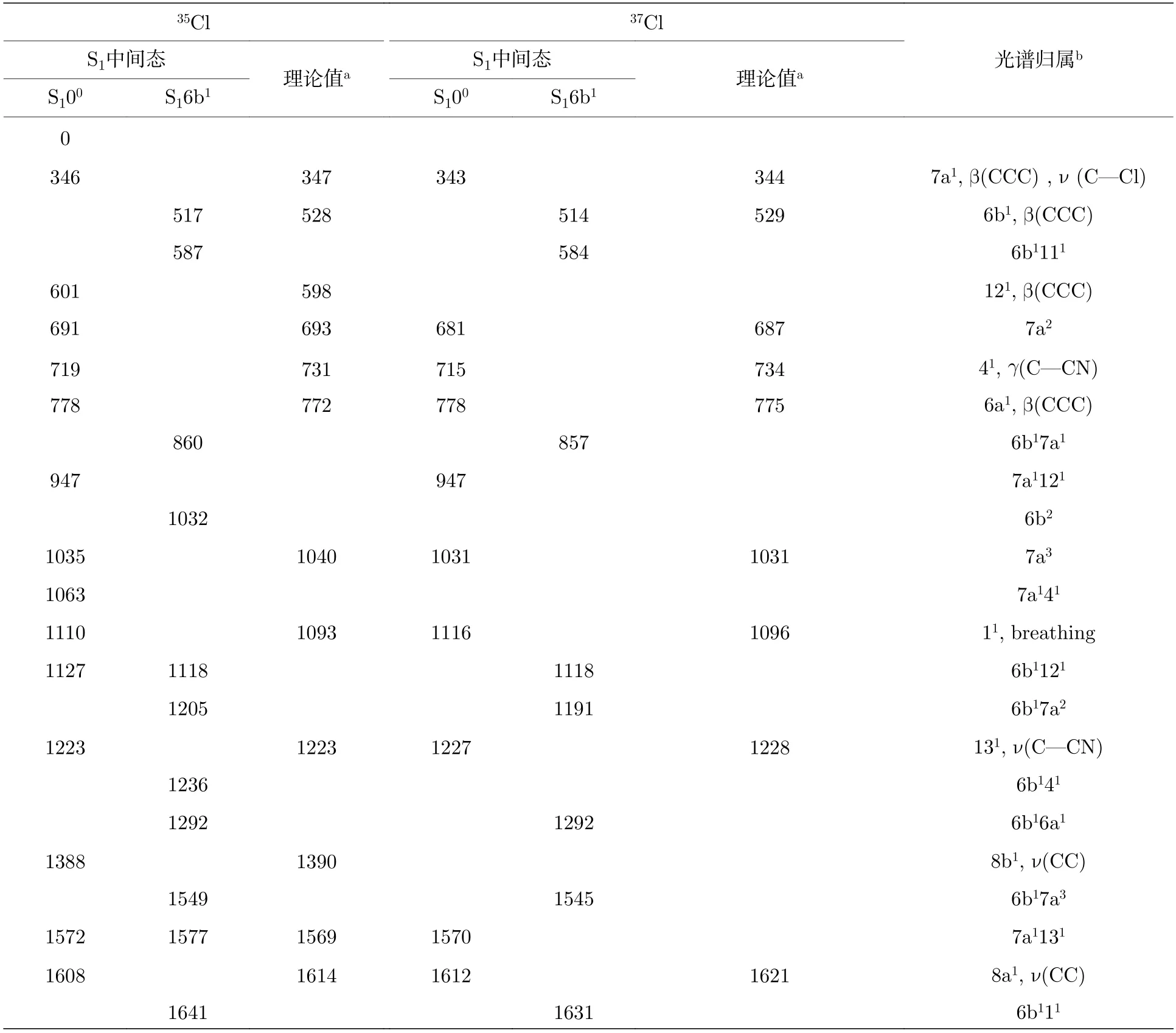
表2 对氯苯腈分子35Cl 和37Cl 同位素离子基态D0 的振动频率及光谱归属a(单位:cm–1)Table 2.The measured vibrational frequencies and assignments in the MATI spectra for the D0 state of 35Cl and 37Cl isotopomers of p-chlorobenzonitrilea (unit:cm–1).
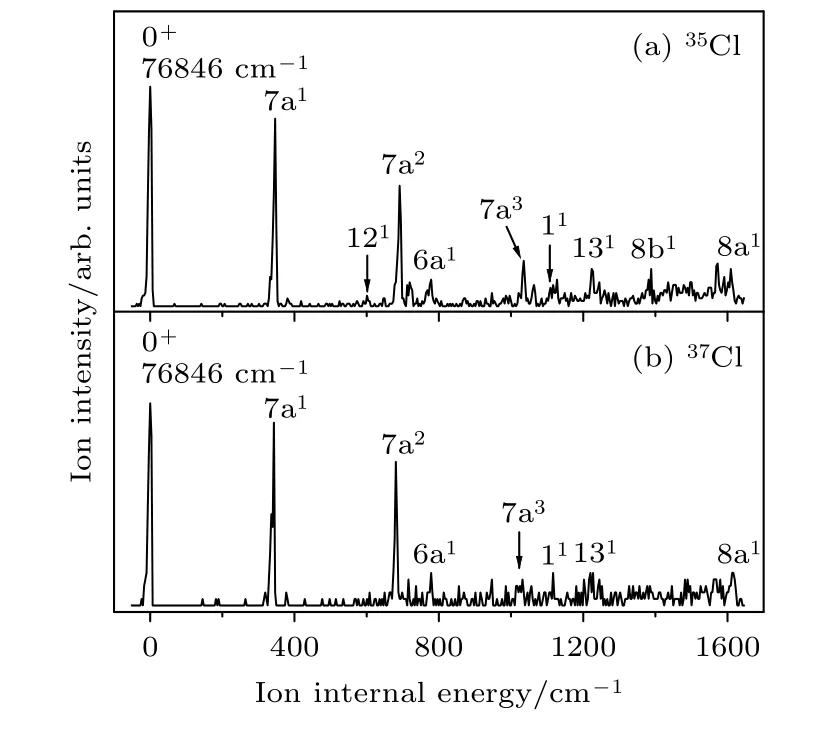
图3 经过对氯苯腈分子35Cl 同位素中间态S100 (a)和37Cl 同位素中间态S100 (b)的MATI 光谱,横坐标是相对对氯苯腈分子电离能的偏移值Fig.3.MATI spectra of the 35Cl (a) and 37Cl (b) isotopomers of p-chlorobenzonitrile via the S100 intermediate state.
图4(a)展示了对氯苯腈35Cl 同位素经过中间态S16b1的MATI 光谱,最强振动峰517 cm–1归属为振动模式6b,表明离子态最强的振动模式与S1中间态相同,满足倾向规则 Δυ=0,类似的特征在对氯苯酚、对氯苯甲醚[29,30]的MATI 光谱中也可以观测到.因此,MATI 光谱上的大多数振动峰可以指认为6b 与其它模的组合振动.振动频率为860 cm–1,1118 cm–1,1205 cm–1,1292 cm–1和1641 cm–1较强的振动峰分别归属为振动模式6b 和其它模式的组合振动6b17a1,6b1121,6b17a2,6b16a1和6b111.图4(b)为对氯苯腈分子37Cl 同位素经过中间态S16b1的MATI 光谱,振动光谱特征与35Cl同位素的MATI 光谱相似.
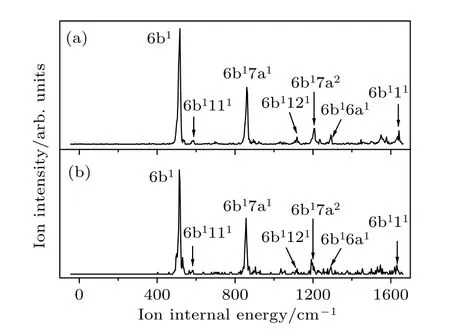
图4 经过对氯苯腈分子35Cl 同位素中间态S16b1 (a) 和37Cl 同位素中间态S16b1 (b) 的MATI 光谱,横坐标是相对对氯苯腈分子电离能的偏移值Fig.4.MATI spectra of the 35Cl (a) and 37Cl (b) isotopomers of p-chlorobenzonitrile via the S16b1 intermediate state.
4 讨论
4.1 对氯苯腈分子结构和振动频率
在S1← S0电子激发和D0← S1电离过程中,对氯苯腈分子结构会发生相应的变化.通过B3-LYP/aug-cc-pVDZ 和 TD-B3LYP/aug-cc-pVDZ方法优化了对氯苯腈分子在中性基态S0、第一电子激发态S1和离子基态D0的分子结构,表3 列出了理论计算和晶体衍射实验[31]的结构参数,分子结构中的原子编号如图1 所示.比较对氯苯腈分子基态S0理论计算的键长、键角,与实验值非常吻合,表明分子结构优化采用的方法是合理的.计算结果中,C8-N9的键长比实验值大0.053 Å,这个误差主要源于对氯苯腈晶体内分子间氰基与氯原子的相互作用[31].比较基态S0与激发态S1的分子结构,在电子激发过程中,对氯苯腈分子苯环上所有六个C-C 键的键长增大,键角C3C4C5和C6C1C2增大,表明苯环发生了扩张,而苯环与氰基和氯原子之间的C1-C8键和C4-Cl7键的键长减小.根据TDDFT 计算,对氯苯腈分子第一电子激发态S1具有ππ* 特征,S1← S0电子跃迁主要源于LUMO ← HOMO 的贡献,分子的电子密度发生了变化,苯环的电子密度增加,而取代基氰基和氯原子电子密度减小,从而引起了相应的激发态分子结构变化.同时,在分子激发态振动光谱中,大多数的活性振动模式9b,7a,6b,6a,1 与苯环和取代基的振动有关,恰好与激发态分子结构变化相对应.在对氯苯酚[24]、对氯苯甲醚[26]、对氟苯腈[32]分子中,也发现了相似的结构变化.
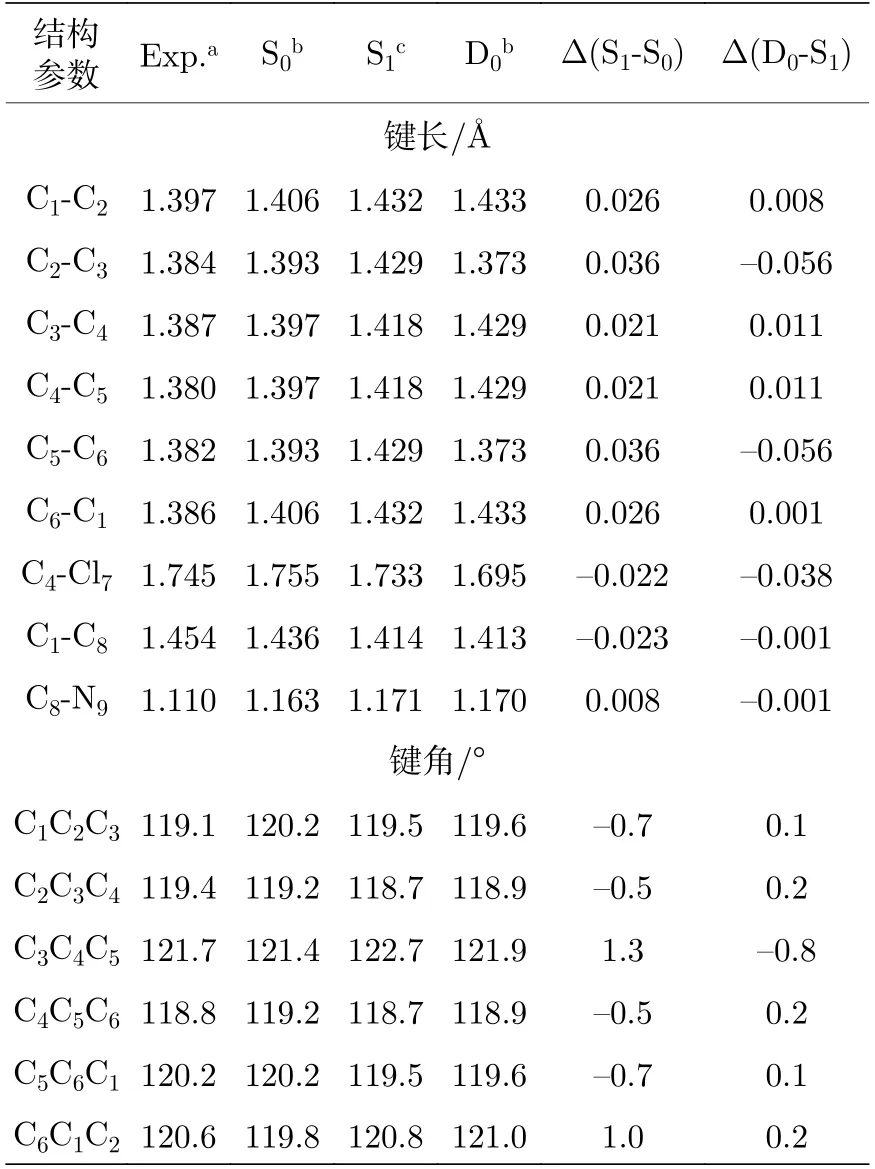
表3 对氯苯腈分子在电子基态、激发态和离子基态的基本结构参数Table 3.Geometrical parameters of p-chlorobenzonitrile in its electronic ground,first excited and cationic ground states.
在对氯苯腈分子的MATI 光谱中,存在一个Δυ=0的振动特征,说明电离过程中分子整体结构变化相对较小,即离子基态的分子结构与激发态的分子结构相似.根据自然布居分析计算,对氯苯腈分子D0← S1电离过程中,苯环上失去一个π*电子,苯环与取代基氰基和氯原子的电子密度减小,因此,C2-C3,C5-C6和C4-Cl7键的键长缩短(见表3),表明整个苯环结构发生了小的收缩,与激发态S1结构比较,离子基态D0苯环整体结构变化仅为1%.对于对氯苯腈35Cl 同位素,离子基态D0时,振动模式7a,6b,6a,1 的振动频率分别为346 cm–1,517 cm–1,778 cm–1和1110 cm–1,激发态S1时,振动频率分别为310 cm–1,504 cm–1,739 cm–1和1067 cm–1,这些苯环平面内β(CCC)弯曲振动在离子基态的振动频率明显高于激发态的振动频率.同样在对氯苯腈37Cl 同位素中,离子基态β(CCC)弯曲振动的振动频率也出现明显的蓝移,表明离子基态时,苯环上化学键的强度大于激发态的强度.
在激发态S1时,对氯苯腈分子35Cl 和37Cl 同位素相同振动模式的振动频率基本相同,说明氯原子的同位素效应对激发态的振动影响很小.这是由于同位素所含电荷相同、电荷分布和静电力场分布非常接近,它们质量数相差非常小引起的.但是,在电离过程中,C4-Cl7键的键长从激发态的1.733 Å减小到离子态的1.695 Å,进一步缩短,表明苯环与氯原子的相互作用变大,引起了两种同位素离子基态振动的频率差异增大.在离子基态时,对氯苯腈分子两种同位素振动模式7a,6b,1,13 的振动频率差异为3—6 cm–1.
4.2 对氯苯腈分子激发能和电离能
苯衍生物分子中,苯环与取代基的相互作用有共轭效应(π 轨道)和诱导效应(σ 键),这两种相互作用引起了苯衍生物分子电子态的能量降低.对于一个从低能级到高能级的电子跃迁,当末态电子态能量降低的大小高于初态电子态能量降低的大小时,跃迁能发生红移,反之,跃迁能蓝移[25].表4 列出了苯酚、苯甲醚、苯胺、苯腈及其衍生物分子的跃迁能[23,29−30,33−38].对甲基苯腈、对氨基苯腈分子的激发能E1相对苯腈的激发能E1发生了红移;同样,对氯苯酚、对氯苯甲醚、对氯苯胺、对氯苯腈分子的激发能E1相对苯酚、苯甲醚、苯胺、苯腈的激发能E1分别红移了1537 cm–1,1524 cm–1,1456 cm–1和700 cm–1,说明S1← S0电子激发过程中,取代基Cl 原子、甲基CH3、氨基NH2与苯环的共轭相互作用增强,主要是因为电子跃迁后苯环扩张,苯环上π 轨道电子与取代基的重叠增加引起的.
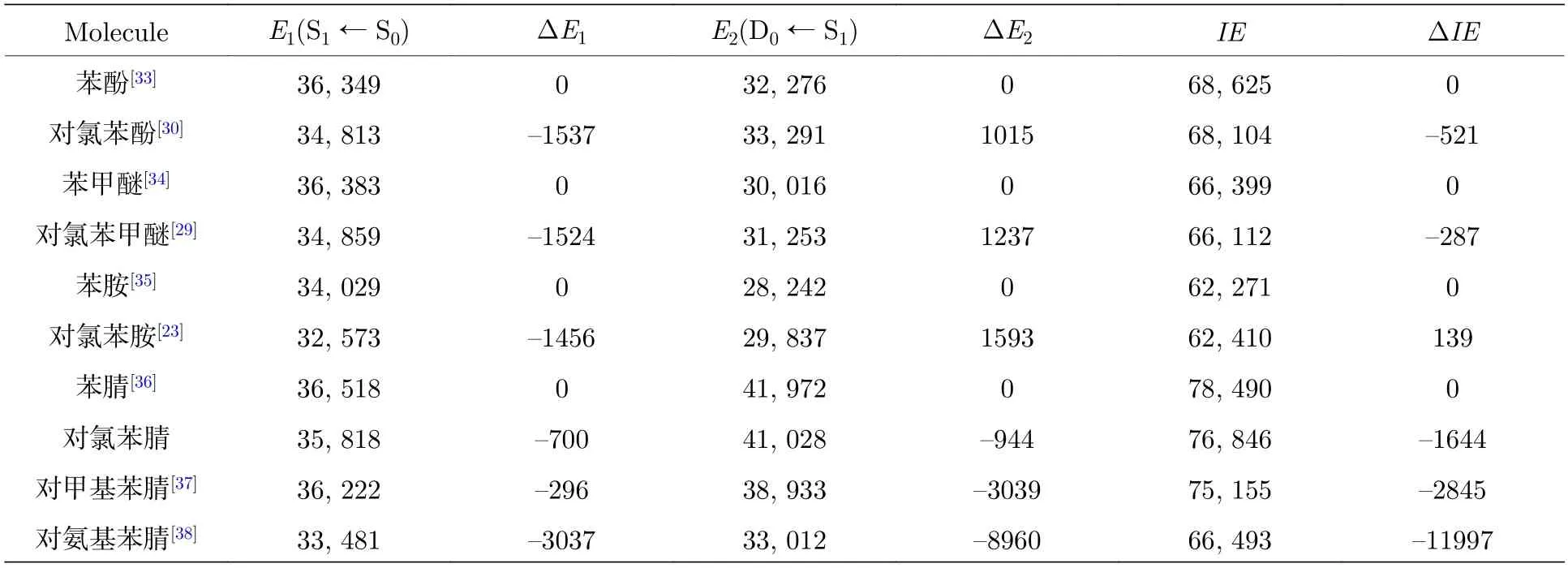
表4 苯酚、苯甲醚、苯胺、苯腈及其衍生物分子的跃迁能(单位:cm–1) aTable 4.The transition energies (cm–1) of phenol,anisole,aniline,benzonitrile and their derivatives.a.
对于D0← S1跃迁能E2,对氯苯酚、对氯苯甲醚、对氯苯胺的跃迁能E2相对苯酚、苯甲醚、苯胺的跃迁能E2发生了蓝移.在D0← S1电离过程中分子失去一个π*电子,跃迁能E2的蓝移说明了取代基Cl 原子表现出吸电子的作用,引起苯环周围电子密度的减小.而对氯苯腈、对甲基苯腈、对氨基苯腈的跃迁能E2相对苯腈的跃迁能E2发生了红移,表明取代基Cl 原子与甲基CH3、氨基NH2具有相同的供电子作用,引起了苯环周围电子密度的增加.因此,不同的苯衍生物分子中,取代基Cl原子表现出了两种不同的电子诱导作用,这主要是由于受到苯衍生物分子中其他取代基的影响.在对氯苯酚、对氯苯甲醚、对氯苯胺分子中,另一个取代基羟基OH、甲氧基OCH3、氨基NH2表现出供电子属性,而在对氯苯腈分子中,另一个取代基氰基CN 表现出比Cl 原子更强的吸电子能力,引起苯环与Cl 原子电子密度的减小,从而诱导了Cl 原子供电子作用.在对氟苯酚、对氟苯甲醚、对氟苯腈分子中[29,32,39],取代基F 原子也表现出了类似的电子诱导作用.
5 结论
本文利用双色共振双光子电离和质量分辨阈值电离光谱技术,测量了对氯苯腈分子第一电子激发态S1和离子基态D0的振动光谱,确定了对氯苯腈分子S1← S0电子跃迁的激发能为35818 ± 2 cm–1,精确的绝热电离能为76846 ± 5 cm–1(9.52766 ±0.0006 eV).对氯苯腈分子35Cl 和37Cl两种同位素有相同的激发能和电离能以及相似的振动特征,第一电子激发态S1和离子基态D0的活性振动大多数是苯环平面内的弯曲振动.在S1← S0电子激发过程中,苯环的电子密度增加,苯环发生了扩张,取代基Cl 原子与苯环的共轭相互作用增强;在D0← S1电离过程中,苯环上失去一个π*电子,分子整体结构发生了小的收缩.对氯苯腈的跃迁能E2相对苯腈的跃迁能E2发生了红移,取代基Cl 原子表现出供电子作用,引起了苯环周围电子密度的增加.
