高分子水凝胶表面蛋白质的吸附行为研究
2022-05-27赵新一宋贺明官小玉王任其陈咏梅
赵新一, 董 坤, 宋贺明, 官小玉, 王任其, 陈咏梅*
(1.陕西科技大学 轻工科学与工程学院, 陕西 西安 710021; 2.西安交通大学 化学学院, 陕西 西安 710049)
0 引言
高分子水凝胶生物材料在应用过程中不可避免地会与人体体液或组织接触而发生蛋白质吸附,因此研究高分子水凝胶材料表面蛋白质吸附行为对于评价其生物相容性、细胞亲和性等性能尤为重要.
生物化学法和现代仪器分析法是目前研究蛋白质吸附的主要方法,如光波导模式光谱[1-3]、椭圆偏振仪[1-3]、全内反射荧光光谱法[4,5]等,具有较高的灵敏性和准确度,可实时动态检测,但得到的信息比较单一,只能反映蛋白质吸附的某一个特性.频率-耗散联用石英晶体微天平(QCM-D)技术能获得蛋白质吸附的多种信息,可动态检测蛋白质在生物材料表面的吸附质量和厚度[1-3],并反映蛋白质的构象变化[6]以及蛋白质在生物材料表面吸附后的黏弹性变化[7]等信息.它利用石英晶体的压电效应而建立起石英共振频率与吸附质量的关系.其应用基础是德国科学家Guenter Sauerbrey提出的Sauerbrey公式[8-10],该公式利用简单的线性关系表述了石英晶体表面吸附质量和频率变化之间的关系.
(1)
式(1)中:Δm为石英微晶天平工作电极表面的沉积物质量变化,可代表蛋白质吸附量的变化.ΔFn是晶体振动频率的变化,可根据其变化直观反映蛋白质吸附的过程.
(2)
式(2)中:耗散因子(D)是指当驱动石英晶体振荡的电路断开后,晶体频率降低到0的时间快慢.间接反映蛋白质的吸附量,即吸附量越多,D值越大.Glost是指晶体在一次振荡周期中能量损耗,Gstore是指晶体在一次振荡周期中存储的全部能量.
在高分子薄膜/液相吸附界面研究体系中,高分子薄膜以及被吸附物质的质量、黏弹性、结构、含水量等因素都包含在QCM-D数据处理提供的频率变化和耗散因子两个参数中.将水凝胶材料成功修饰于石英晶片表面是使用QCM-D技术监测水凝胶吸附蛋白质的前提.目前报道的方法有两种:一是物理吸附方法[11-14],将旋转涂覆于石英晶片表面的亲水性高分子薄膜烘干后近似看作水凝胶,该方法中石英晶片能够反复利用,节约实验成本,但构成该薄膜的高分子链没有被交联形成三维水凝胶网络结构,从而会缺失水凝胶网络结构对蛋白质吸附性能的影响;二是化学接枝方法[15-20],即通过共价键反应在石英晶片表面修饰活性双键,然后将亲水性单体表面原位引发接枝于石英晶片表面形成水凝胶网络结构,该方法虽然保持了水凝胶三维网络结构,但石英晶片不能反复利用,增加了实验成本.故而,尝试通过微凝胶吸附方法在石英晶片表面制备一层水凝胶薄膜进而监测微凝胶材料表面蛋白质吸附过程.
1 实验部分
1.1 实验原料
N,N′-二甲基丙烯酰胺,分析纯,TOKYO KASEI公司;2-丙烯酰胺基-2-甲基丙磺酸钠,分析纯,山东省煜化学有限公司;甲基丙烯酰氧乙基三甲基氯化铵,分析纯,Alfa Aesar公司;N,N′-亚甲基双丙烯酰胺,国药集团化学试剂有限公司;α-酮戊二酸,分析纯,上海科丰化学试剂有限公司;牛血清白蛋白(BSA),北京奥博星生物技术有限公司.
1.2 实验仪器
电子天平,BS224S,德国Sartourius公司;紫外灯,365 nm/40 W,Philips公司;搅拌机,MJ-250BP02A,广东美的生活电器制造有限公司;纳米激光粒度仪,Nano ZS 90,MALVERN公司;频率-耗散联用石英微晶天平,Q-SenseE4,瑞典Q-Sense公司;镀金石英晶片,AT切,5MHz,瑞典Q-Sense公司.
1.3 实验方法
1.3.1 高分子水凝胶的制备
三种乙烯类单体通过光引发反应合成高分子水凝胶材料,其中2-丙烯酰胺-2-甲基丙烷磺酸钠(NaAMPS)作为阴离子单体,N,N′-二甲基丙烯酰胺(DMAAm)作为中性单体,甲基丙烯酰氧乙基三甲基氯化铵(METAC)作为阳离子单体.具体操作步骤如下:将单体配制成1 mol/L的水溶液,以单体浓度为参比加入交联剂N,N′-亚甲基双丙烯酰胺(MBAA).待单体和交联剂完全溶解后再以单体浓度为参比加入0.1 mol%引发剂α-酮戊二酸,并通入氮气30 min.将该溶液加入洁净玻璃夹具间,用波长为365 nm紫外光照射12 h.最后将制备的水凝胶材料取出,浸泡在去离子水中溶胀,每天换水2次持续一周,保证水凝胶中未反应的成分被充分置换并达到溶胀平衡.最后浸泡在磷酸缓冲液(PBS)中交换溶胀,每天更换PBS溶液两次,持续三天使其与人体组织保持等渗状态.
1.3.2 微凝胶的制备
将PBS溶液中溶胀平衡的高分子水凝胶材料放于PBS溶液中,搅拌5 min后获得微凝胶溶液.静置24 h待沉析完成后取上层清液,获得稳定的微凝胶溶液.利用纳米激光粒度仪分析微凝胶粒径大小及分布情况.
1.3.3 石英晶片及微凝胶表面蛋白质吸附过程
利用QCM-D技术监测石英晶片表面蛋白质吸附过程.在37 ℃(体温)、150 uL /min进样速率下,通入PBS溶液,待石英晶片振动频率稳定后,将该频率记录为测试零点.通入PBS溶液3 min后更换通入BSA溶液,待石英晶片振动频率稳定后,依次从低浓度到高浓度更换BSA溶液,记录石英晶片振动频率变化(ΔFn)及耗散因子变化(ΔDn)随时间的变化,n取值为1、3、5、7、9和11.选取ΔF3及ΔD3作为归一化值,用以数据处理及结果分析.
利用QCM-D技术监测三种微凝胶材料表面蛋白质吸附过程.在37 ℃(体温)、150 uL/min进样速率下,通入PBS溶液,待石英晶片振动频率稳定后,将该频率记录为测试零点.通入PBS溶液3 min后更换通入微凝胶溶液,待石英晶片振动频率稳定后,依次从低浓度到高浓度更换通入BSA溶液,记录石英晶片振动频率变化(ΔFn)及耗散因子变化(ΔDn)稳定随时间的变化,n取值为1、3、5、7、9和11.选取ΔF3及ΔD3作为归一化值用以数据处理及结果分析.
2 结果与讨论
2.1 三种高分子微凝胶的粒径分布
图1、2、3分别为不同交联密度的PMETAC、PNaAMPS及PDMAAm三种高分子微凝胶粒径分布图,其横坐标为粒径大小,纵坐标为微凝胶溶液中某一粒径微凝胶数所占整个微凝胶总数量的比例.从图中可以看出,三种微凝胶不同交联密度粒径分布相对较为均匀,PMETAC微凝胶粒径分布在600~700 nm,PDMAAm微凝胶粒径分布在800~1 000 nm,PNaAMPS微凝胶粒径分布在400~700 nm.由于三种微凝胶材料粒径分布均匀,因此在后期物理吸附研究中不考虑其粒径对吸附性能的影响.
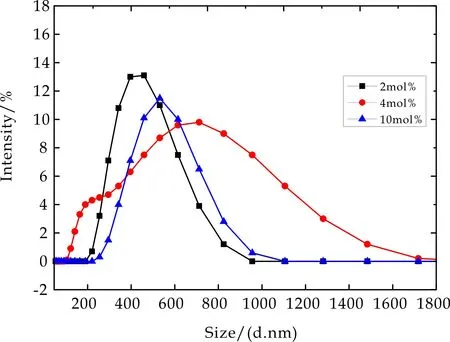
图2 不同交联密度PNaAMPS微凝胶粒径分布
2.2 石英晶片表面蛋白质吸附过程
利用QCM-D技术监测三种微凝胶材料表面蛋白质吸附过程,必须确定洁净石英晶片表面蛋白质吸附情况,并在计算微凝胶表面BSA吸附量时扣除洁净晶片表面吸附量.图4为石英晶片表面吸附BSA过程中,石英晶片振动频率ΔF3和相应耗散因子ΔD3随时间变化曲线.由图可以看出,随着BSA吸附时间的增加,ΔF3略微降低,ΔD3略微增大,最后达到平衡,且随着BSA浓度的提高,ΔF3及ΔD3的变化均增大.说明石英晶片表面有微量的蛋白吸附.
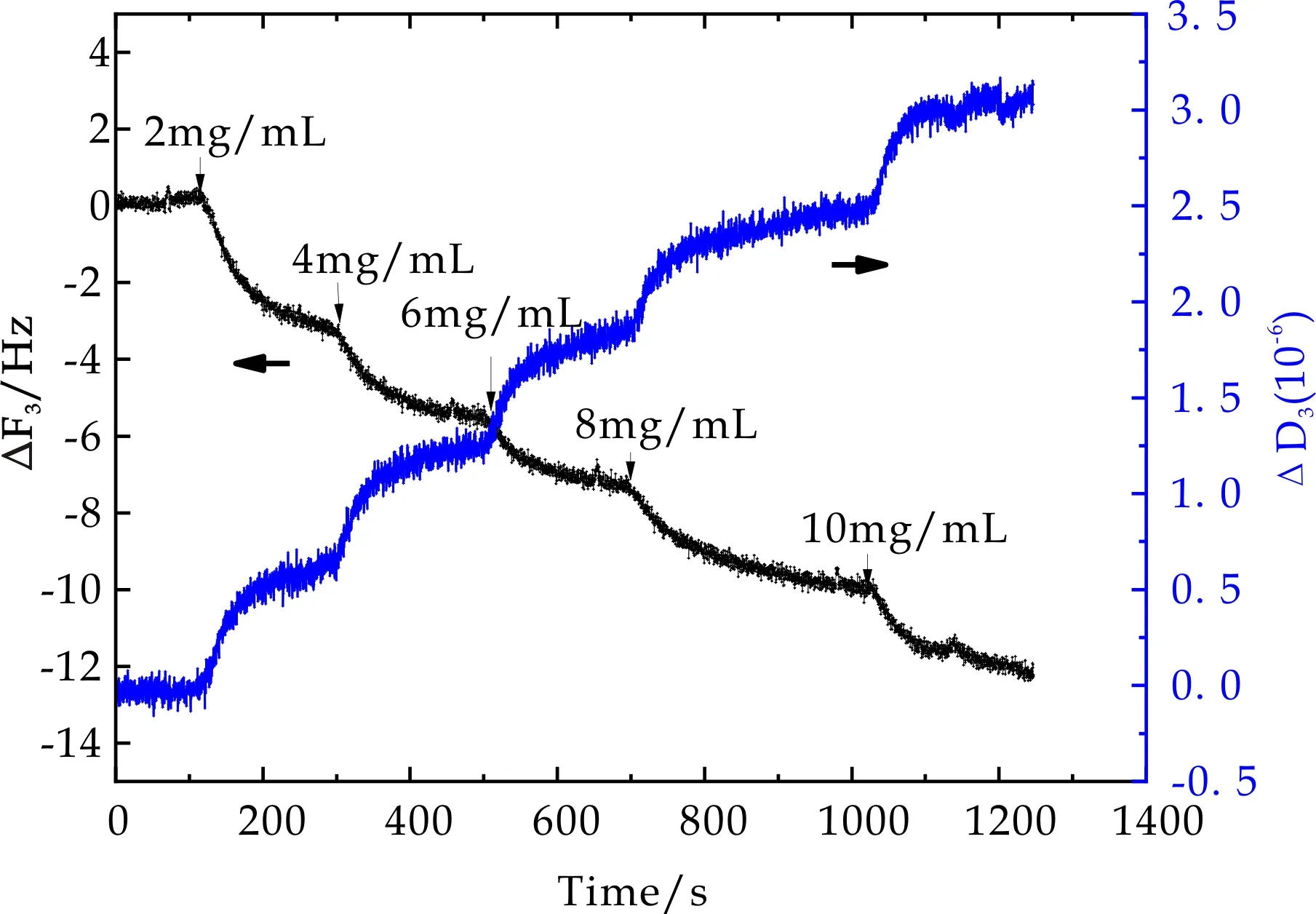
图4 洁净晶片表面蛋白质吸附过程频率及耗散因子随时间变化曲线
2.3 微凝胶表面蛋白质吸附过程
2.3.1 PMETAC微凝胶表面蛋白质吸附过程
以相同实验方法获得不同交联密度的正电荷PMETAC微凝胶,其表面BSA吸附过程中,石英晶片振动频率ΔF3和相应耗散因子ΔD3随时间变化曲线如图5~7所示.从图中可以看出,通入微凝胶溶液后ΔF3随时间增加不断降低,ΔD3随时间增加不断增大,最终达到吸附平衡,表明微凝胶颗粒能够在振子表面发生物理吸附.当微凝胶吸附达到平衡后,再用PBS冲洗时,ΔF3和ΔD3的变化均非常微小,由此推断,微凝胶在石英晶片表面的吸附非常稳定.当通入2 mg/mL BSA溶液后,ΔF3随时间增加显著降低,ΔD3随时间增加而增大,最终达到吸附平衡.此外,不同BSA浓度环境下,虽然ΔF3随时间增加不断降低,ΔD3随时间增加不断增大,但在BSA浓度从4 mg/mL升高至10 mg/mL过程中,每次BSA浓度的提高所引起的ΔF3和ΔD3的变化趋势相对2 mg/mL时变化非常缓和.不同交联密度的PMETAC微凝胶表面BSA吸附量近似.
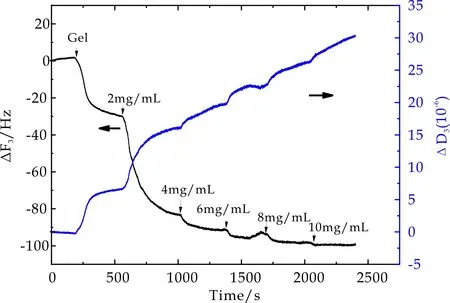
图5 2mol%PMETAC在BSA吸附过程中频率及耗散因子随时间变化曲线
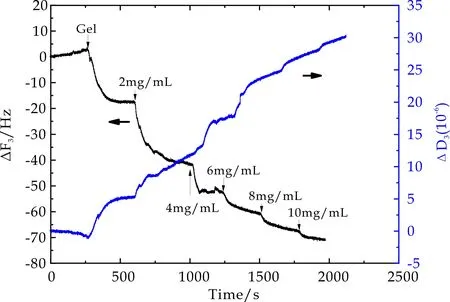
图6 4mol%PMETAC在BSA吸附过程中频率及耗散因子随时间变化曲线
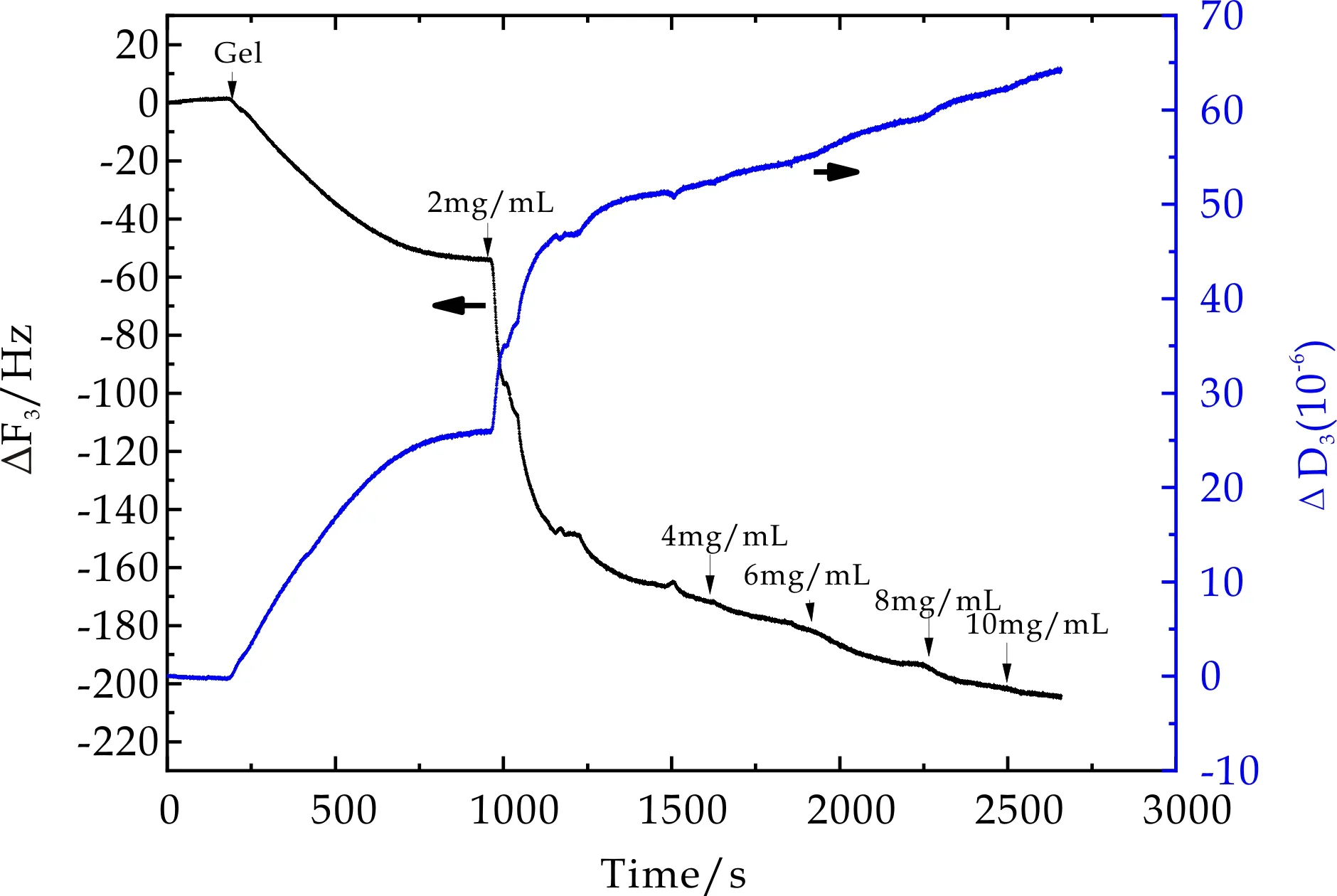
图7 10mol%PMETAC在BSA吸附过程中频率及耗散因子随时间变化曲线
2.3.2 PDMAAm微凝胶表面蛋白质吸附过程
以相同实验方法获得不同交联密度的中性PDMAAm微凝胶,其表面BSA吸附过程中,石英晶片振动频率ΔF3和相应耗散因子ΔD3随时间变化曲线如图8~10所示.从图中可以看出,随着微凝胶溶液和BSA溶液的通入,ΔF3随时间增加不断降低,ΔD3随时间增加不断增大,并最终达到稳定值.表明微凝胶颗粒能够在振子表面发生物理吸附.当微凝胶吸附达到平衡后,再用PBS冲洗时,ΔF3和ΔD3的变化均非常微小,由此推断,微凝胶在石英晶片表面的吸附非常稳定.当通入2 mg/mL BSA溶液后,ΔF3随时间增加显著降低,ΔD3随时间增加而增大,最终达到吸附平衡.此外,不同BSA浓度环境下,虽然ΔF3随时间增加不断降低,ΔD3随时间增加不断增大,但是随着BSA浓度从4 mg/mL升高至10 mg/mL过程中,每次BSA浓度的提高所引起的ΔF3和ΔD3的变化趋势相对2 mg/mL时的变化非常缓和.PDMAAm微凝胶表面BSA吸附过程与PMETAC微凝胶相同,但ΔF变化较小,根据Sauerbrey公式可得,PDMAAm表面的BSA吸附量小于PMETAC.不同交联密度的PDMAAm微凝胶表面BSA吸附量近似.
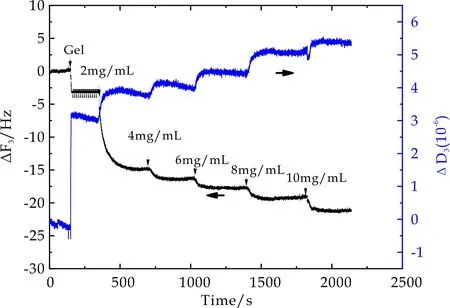
图8 2mol%PDMAAm在BSA吸附过程中频率及耗散因子随时间变化曲线
2.3.3 PNaAMPS微凝胶表面蛋白质吸附过程
以相同实验方法获得不同交联密度的负电荷PNaAMPS微凝胶,其表面BSA吸附过程中,PNaAMPS微凝胶溶液的通入并未使石英晶片振动频率ΔF3和耗散因子ΔD3发生变化.因此推断,石英晶片表面羟基在水中电离后与PNaAMPS表面带有的负电荷基团发生电荷排斥作用,使PNaAMPS微凝胶颗粒未在石英晶片表面发生吸附.为获得PNaAMPS微凝胶表面BSA吸附数据,我们通过层层自组装的方法在石英晶片表面形成一层稳定的PMETAC微凝胶膜,用PBS进行冲洗,再通入PNaAMPS微凝胶溶液,获得一层PNaAMPS微凝胶薄膜,进而监测其表面BSA吸附情况.
图11~13为PNaAMPS薄膜表面吸附BSA过程中石英晶片振动频率ΔF3和耗散因子ΔD3随时间的变化曲线.当石英晶片表面PMETAC(2 mol%)吸附平衡后,用PBS冲洗过程中ΔF3和ΔD3并不发生变化,当通入PNaAMPS微凝胶溶液后,ΔF3显著降低,ΔD3显著增高.对比PMETAC及PDMAAm微凝胶吸附过程可知,PNaAMPS微凝胶颗粒在PMETAC薄膜表面发生了吸附作用,即利用异种电荷间相互吸引作用在石英晶片表面通过层层自组装可以形成一层PNaAMPS微凝胶薄膜,并且该薄膜非常稳定(PBS冲洗未发生频率及耗散因子变化),从而保证了QCM-D技术监测BSA在PNaAMPS表面吸附情况的可行性.
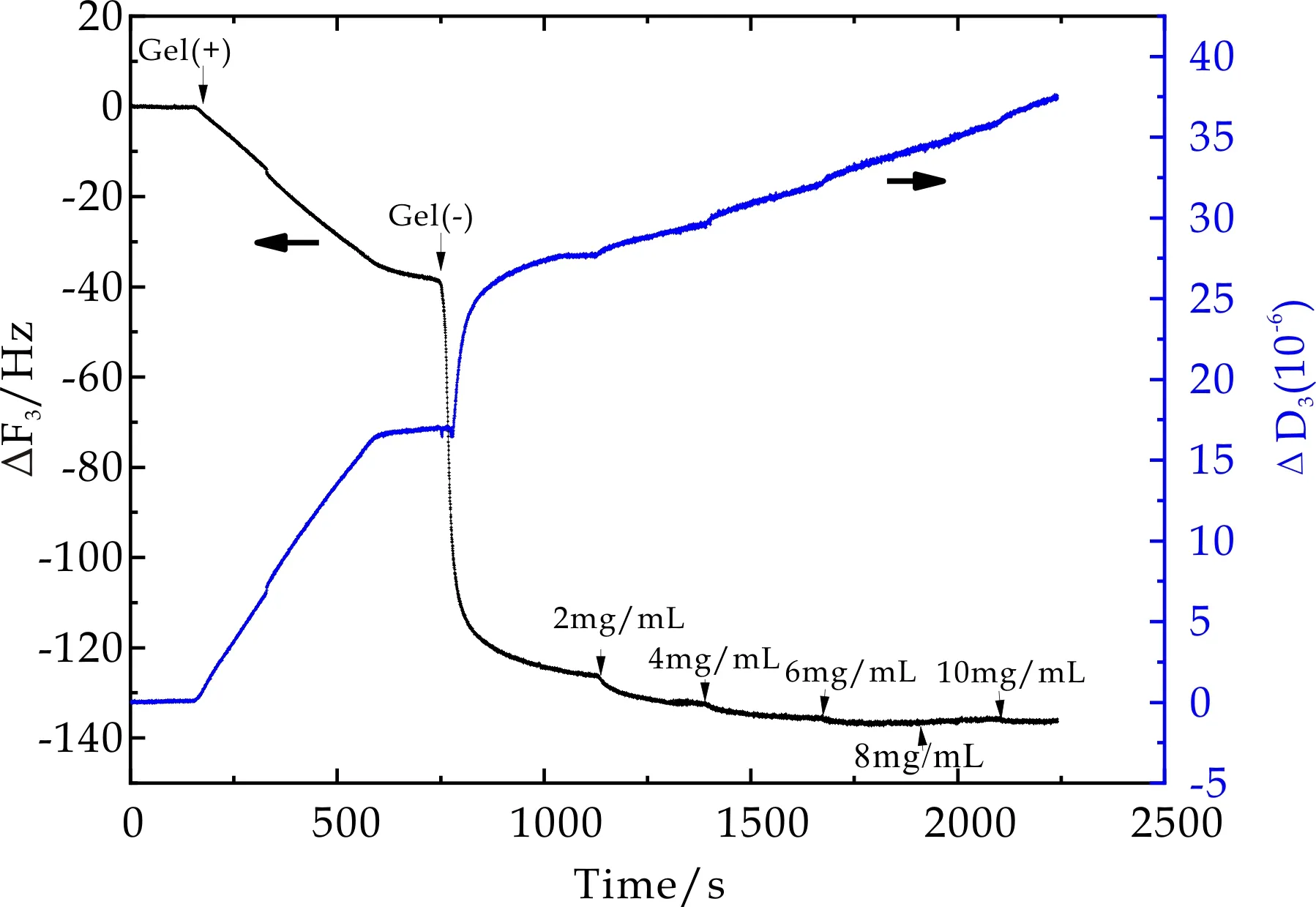
图11 2mol%PNaAMPS在BSA吸附过程中频率及耗散因子随时间变化曲线
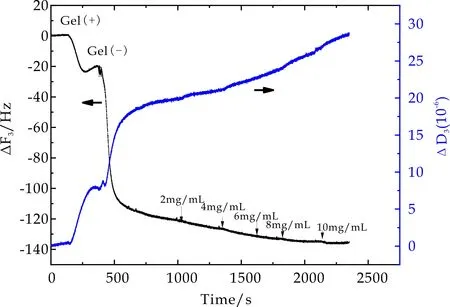
图12 4mol%PNaAMPS在BSA吸附过程中频率及耗散因子随时间变化曲线
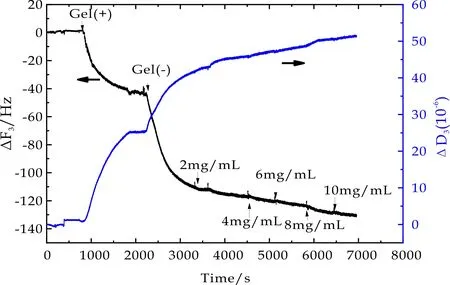
图13 10mol%PNaAMPS在BSA吸附过程中频率及耗散因子随时间变化曲线
随着BSA溶液的通入,ΔF3随时间增加不断降低,ΔD3随时间增加不断增大.相对于PMETAC和PDMAAm微凝胶表面BSA吸附变化,PNaAMPS微凝胶表面BSA吸附过程中ΔF3及ΔD3变化量非常小,推测可能BSA在PNaAMPS表面吸附量较低.不同交联密度的PNaAMPS微凝胶表面BSA吸附量近似.
2.4 微凝胶表面BSA吸附性能分析
|ΔD/ΔF|描述振子表面的表观吸附层的粘弹性,其值越小则表示该吸附层的刚性越好.图14为洁净晶片表面BSA吸附过程中D-F曲线图.从图中可以看出,随着ΔF3的降低,ΔD3不断增加,且呈简单线性变化关系,说明在洁净石英晶片表面BSA有微量吸附.
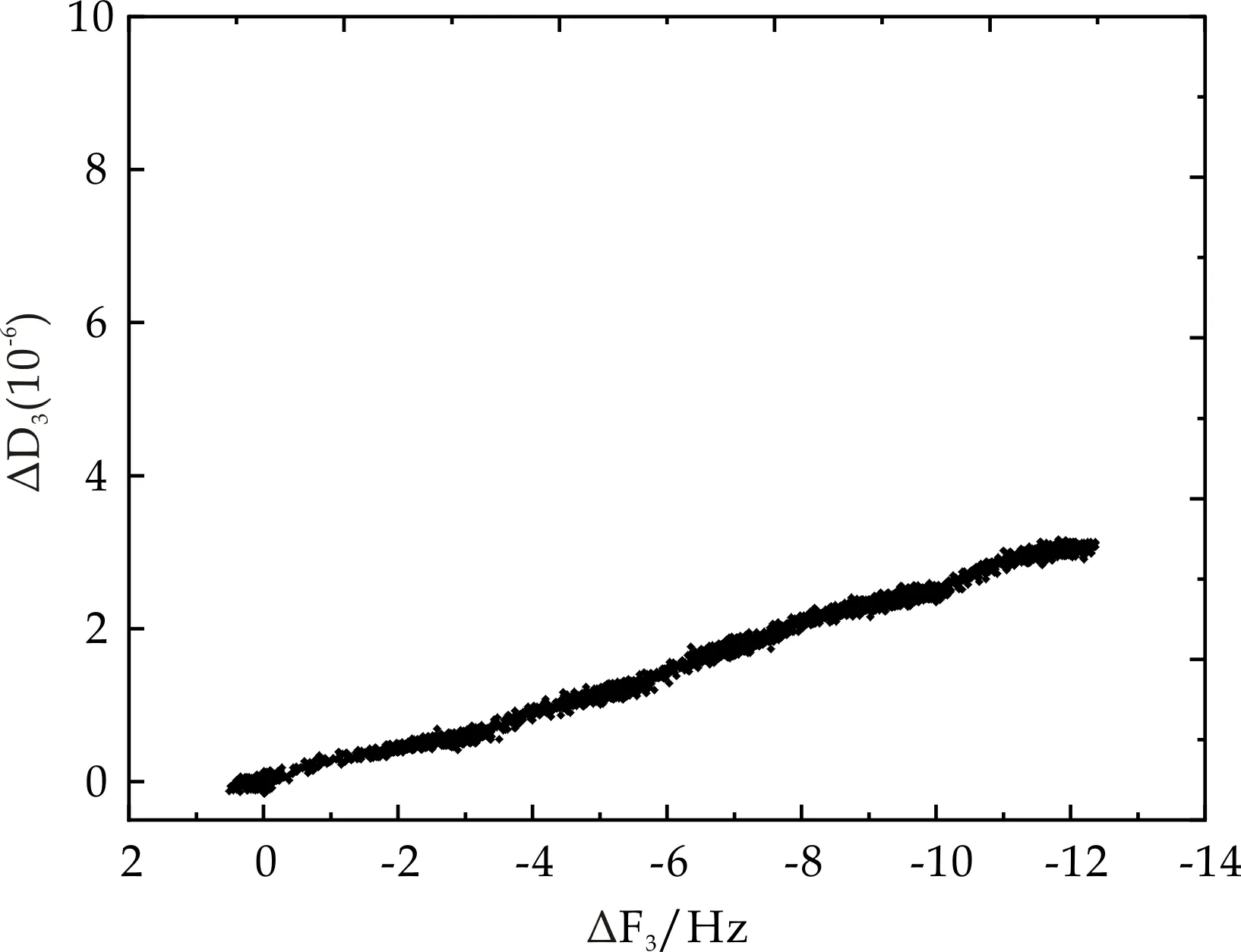
图14 洁净晶片表面耗散因子随频率变化趋势
相同的处理方法,对三种微凝胶表面BSA吸附过程中的|ΔD3/ΔF3|关系进行作图,从而获得其吸附性能初步分析.图15~17所示分别为PMETAC、PDMAAm及PNaAMPS三种微凝胶吸附中耗散因子随频率变化曲线.
从图15和图16可以看出,当BSA浓度为2 mg/mL时,PMETAC及PDMAAm两种微凝胶表面BSA吸附过程中ΔD3随ΔF3呈线性变化趋势,且其线性变化斜率小于相应微凝胶吸附过程中斜率,该现象表明在2 mg/mL BSA液相环境下,BSA在PMETAC及PDMAAm两种微凝胶表面吸附形成一层更致密薄膜.而当BSA浓度再次提高,ΔD3随ΔF3变化剧烈增高,已经不呈现线性关系,即当BSA浓度大于2 mg/mL时,耗散因子的增加远大于频率变化,即BSA浓度的提高仅仅增大了系统粘弹性,而吸附质量并没有多大改变,由此推测,PMETAC及PDMAAm两种微凝胶表面在BSA浓度为2 mg/mL时即发生饱和吸附.图17中PNaAMPS微凝胶表面BSA吸附过程中ΔD3随ΔF3降低明显增大,说明其材料耗散变化远大于频率变化,即BSA在PNaAMPS表面形成一层松散且有粘弹性的的膜层.
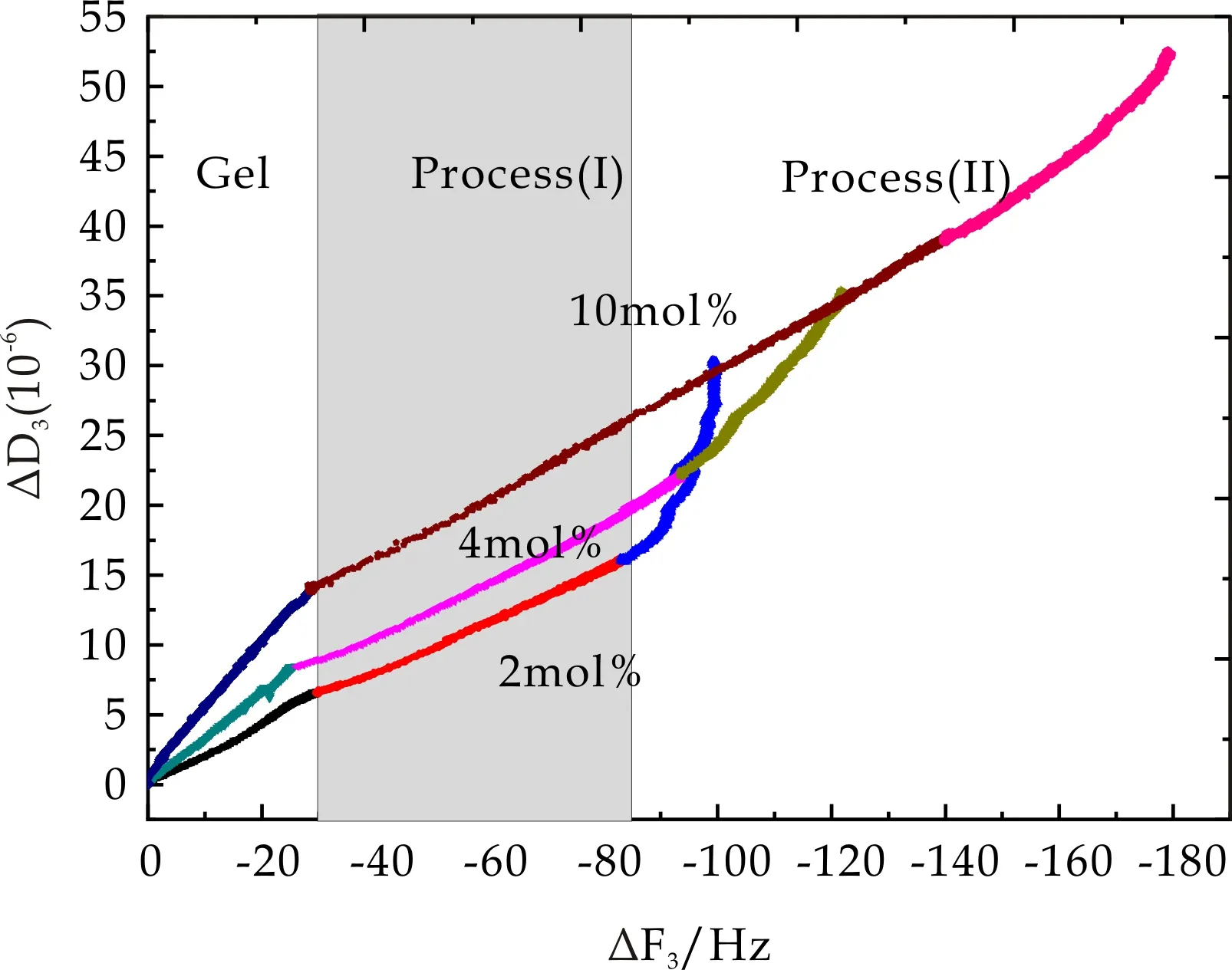
图15 PMETAC微凝胶吸附及其表面BSA吸附过程中耗散因子随频率变化趋势
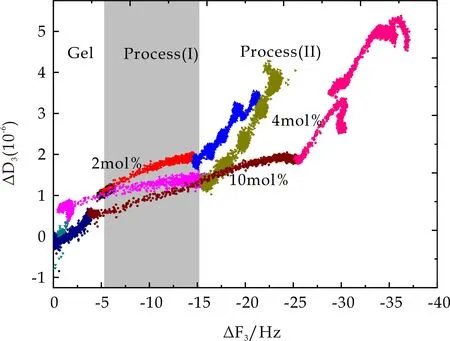
图16 PDMAAm微凝胶吸附及其表面BSA吸附过程中耗散因子随频率变化趋势
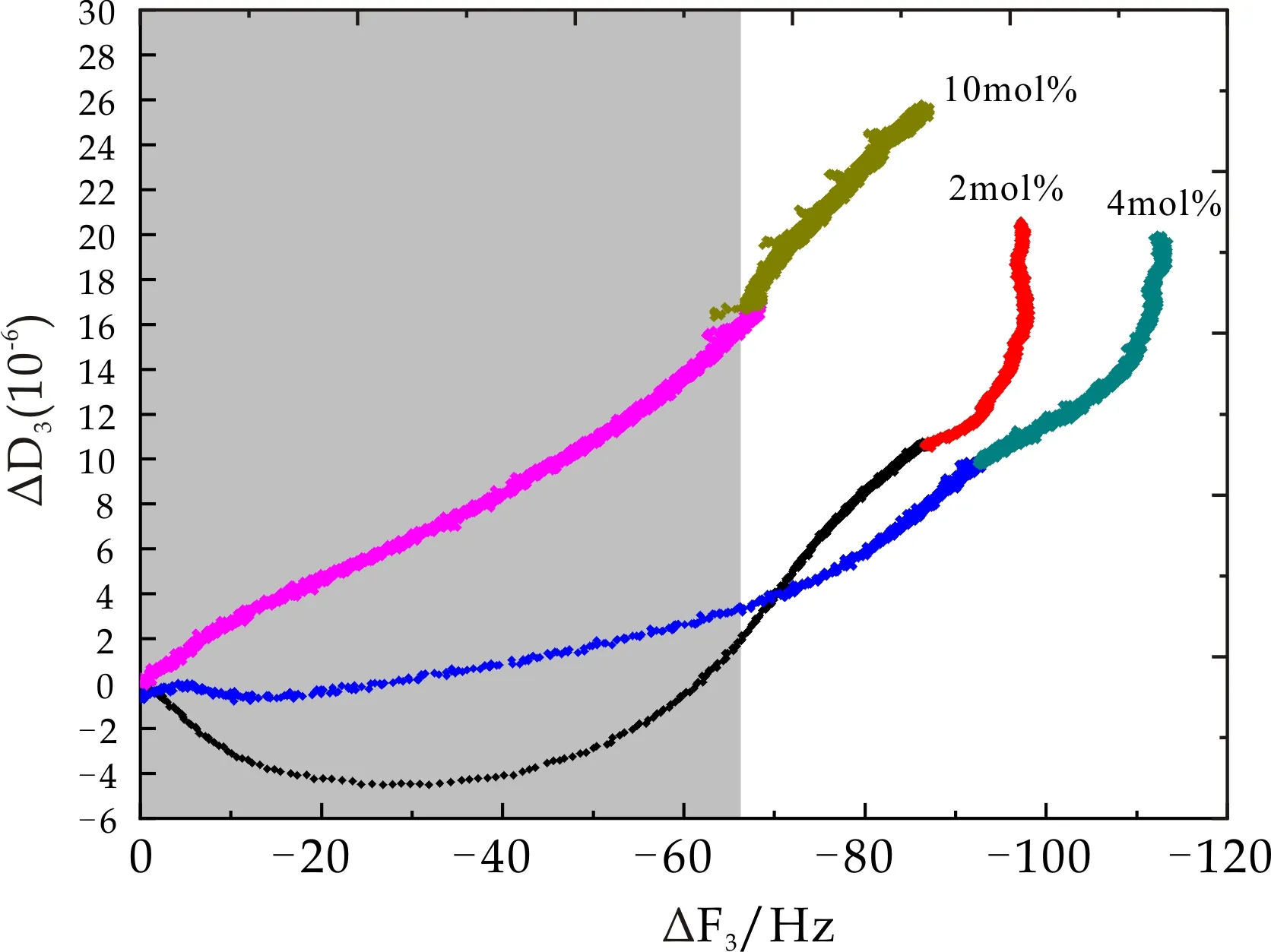
图17 PNaAMPS微凝胶吸附及其表面BSA吸附过程中耗散因子随频率变化趋势
3 结论
发展了微凝胶吸附方法在石英晶片表面获得一层水凝胶薄膜,并采用石英微晶天平技术(QCM-D)分析了带正电荷的PMETAC、中性的PDMAAm及带负电荷的PNaAMPS表面蛋白质吸附行为.得到以下结论:
(1)通过物理分散获得的微凝胶溶液能够在石英晶片表面吸附形成一层致密薄膜,该方法具有切实可行的操作性,为利用QCM-D技术监测水凝胶材料表面蛋白质吸附性能提供了基础.
(2)利用QCM-D技术监测了三种不同电荷微凝胶材料表面BSA吸附行为.单位质量微凝胶表面BSA吸附质量顺序为PDMAAm>PMETAC >PNaAMPS,交联密度并不影响水凝胶材料表面BSA吸附量.
(3)三种微凝胶表面在BSA浓度为2 mg/mL时即达到饱和吸附,BSA浓度的继续提高也仅提高了体系粘弹性.
