磷化法制备NixPy及其复合材料光解水性能
2022-05-26颜瑞孙志超张蒙蒙刘颖雅遇治权王伟王瑶王安杰
颜瑞,孙志超,张蒙蒙,刘颖雅,遇治权,王伟,王瑶,王安杰
(1 大连理工大学精细化工国家重点实验室,辽宁 大连 116024;2 辽宁省高校石油化工技术与装备重点实验室,辽宁 大连 116024;3 银川能源学院,宁夏 银川 750105)
过渡金属磷化物作为一类优异的催化材料,在加氢精制、光催化、电催化、能量转换与储存等领域表现出优异的性能。磷化镍(NiP)是其中最具代表性的一类磷化物,具有多种化学计量形式,包括NiP、NiP、NiP、NiP、NiP 和NiP等结构,其催化性质敏感地取决于其晶相和电子结构。比如,NiP在电催化分解水制氢反应中表现出最高的反应性能,NiP 是各种NiP中加氢脱硫的最优活性相,但在加氢脱氧过程中,NiP 会在高温水相条件下逐渐转变为NiP,且NiP表现出优异的加氢活性和高温水相稳定性。因此,研究合成条件对磷化镍晶相形成的影响至关重要。
磷化镍的常规制备方法包括程序升温还原磷酸盐法、次/亚磷酸盐歧化法和有机磷液相合成法等。有机磷液相合成法可以制备多种NiP晶相,如Li 等通过液相合成法以Ni 的有机盐为前体、三辛基膦(TOP)为磷源,调控P/Ni 摩尔比、反应温度及时间,成功制备了NiP、NiP、NiP三种晶相。以无机磷化物为磷源制备磷化镍时,很难制备具有高P/Ni 摩尔比的单一NiP晶相,如遇治权通过程序升温还原磷酸盐法,以Ni(NO)和(NH)HPO为前体及磷源组合,改变Ni/P摩尔比制备NiP、NiP、NiP 三种磷化镍晶相。此外,以NiS 与NaHPO、NiCl与NHHPO为前体及磷源组合,通过次/亚磷酸盐歧化法得到的样品均为NiP晶相。因此,研究无机磷源对NiP晶相的影响规律,有助于拓展磷化镍的制备方法和明确磷化镍各晶相间的转化规律。
本文提出了一种红磷磷化的方式用于制备多种NiP晶相。该方法以成本低廉的红磷为磷源,NiP为前体,通过调控合成温度成功制备出NiP、NiP、NiP三种晶相,同时考察了Ni/P 摩尔比、磷化时间、磷化温度、磷化气速、磷源种类等因素对制备晶相的影响。将上述4种NiP与CdS混合制备NiP/CdS 复合材料,研究其光催化分解水产氢反应性能。
1 实验部分
1.1 实验试剂
六水合硝酸镍[Ni(NO)·6HO]、磷酸氢二铵[(NH)HPO]、红磷、次亚磷酸钠(NaHPO·HO)、硝酸镉、硫脲、乙二胺、,二甲基甲酰胺、无水乙醇,均为分析纯,国药化学试剂公司。
1.2 催化剂制备
1.2.1 NiP制备
体相NiP可通过氢气程序升温还原法(H-TPR)制备。取13.2g Ni(NO)·6HO和2.0g(NH)HPO分别溶于适量去离子水中形成澄清溶液,分别记为A和B。在搅拌条件下,将B逐滴滴入A 中,得浅绿色悬浊液。继续搅拌30min后,将悬浊液在电炉上搅拌蒸干,得深绿色黏稠状固体。所得固体在120℃干燥12h,研磨,500℃焙烧3h,得到黑色粉末状前体。将粉末状前体压片、破碎至20~40 目。称取0.5g装入还原钝化炉,在150mL/min H气氛下以2℃/min 升温至400℃,稳定10min后,以1℃/min升温至500℃还原2h,降至室温后,在0.5%O/Ar气氛下钝化处理2h,得到体相NiP。
1.2.2 NiP的制备
取一定量红磷(或次亚磷酸钠)和50mg NiP分别置于U 形管的上下游,并以石英棉将其隔开。以20mL/min 气速通入N吹扫10min 后,采用不同的磷化终温、磷化时间和升温速率和N气速,确保气体均匀通过NiP 床层,制备不同晶相的NiP样品。
1.2.3 NiP/CdS的制备
CdS 通过水热法制备。95mg CdS 粉末分散至10mL,二甲基甲酰胺溶液,超声30min后,加入5mg NiP,搅拌12h,离心分离。固体经多次水洗、乙醇洗后,置于80℃烘箱干燥12h,研磨得到颜色均一的黄绿色粉末,所得样品记为NiP/CdS。
1.3 表征和反应性能评价
1.3.1 表征
催化剂晶体结构由D/MAX-2400型X射线衍射仪(XRD)测定;催化剂元素价态组成由Escalab XI+X 射线光电子能谱仪测定;催化剂光响应范围由UV-550 型紫外分光光度计测定;催化剂载流子分离效率通过F7000型荧光光谱仪进行光致发光光谱(PL)测试,激发波长为350nm,扫描范围400~700nm。
1.3.2 光解水反应性能评价
取2mg 光催化剂装入30mL 石英反应管,加入5mL 0.1mol/L NaS-NaSO超纯水溶液将催化剂分散均匀。放入转子,用胶塞将样品管封口,向样品管内通入氩气30min,排出管内的空气。开启氙灯光源(中教金源,CEL-PF300-T8),使用300W 功率可见光(使用滤光片取波长大于420nm)对催化体系照射4h,光照强度为54mW/cm,光照面积为0.785cm,每小时取200μL 气体通过GC(天美-6890,TCD 检测器,5A 分子筛填充柱)检测H浓度。连续三次实验数据误差小于5%的情况下取平均值,计算材料的产氢速率,并按照式(1)计算表观光量子转化效率(AQE)。

2 结果与讨论
2.1 制备NixPy的影响因素
2.1.1 磷化温度和磷化时间的影响
如图1(a)所示,通过H-TPR 法制备的NiP 样品,其XRD衍射峰出现在2=36.4°、41.7°、42.8°、43.6°、45.3°、46.0°、46.6°、50.6°、52.1°、52.8°、55.4°、75.4° 和75.5°处,与NiP 晶相(JCPDS 74-1384)PDF 卡片一致。当Ni/P 摩尔比为1∶4、磷化时间为2h、磷化气速为20mL/min 时,使用红磷在不同温度下对NiP 进行磷化。当磷化温度为400℃时,所得样品为NiP 和NiP的混合晶相,当磷化温度提高到500℃时,所得样品则表现为纯净的NiP晶相(JCPDS 18-0883),其特征衍射峰位于2=36.1°、40.6°、41.4°、43.9°、45.1°、47.0°、47.8°、49.8°、53.0°和54.0°处,且谱图中无杂峰出现,说明在合成NiP的过程中,NiP 经NiP过渡,逐渐完成向NiP晶相的变化。当磷化温度升高至600℃,样 品XRD 谱 峰 仅 在2=40.8°、44.6°、47.3°、54.2°、54.9° 处 出 现,与NiP 的 衍 射 峰(JCPDS 03-0953)一致,证明此磷化温度下制备的样品为NiP晶相。当磷化温度为800℃时,XRD谱图中出现典型的NiP特征衍射峰(JCPDS 22-11900),分别在2=38.4°、41.6、41.7°、46.9°、48.9°处。上述结果说明,通过改变磷化温度,NiP可以依次被磷 化 为NiP(500℃)、NiP (600℃)、NiP(800℃)三种晶相。由此可知,磷化温度对最终晶相的形成起决定作用,在最终晶相形成前,样品中可能有一种或多种过渡晶相存在。
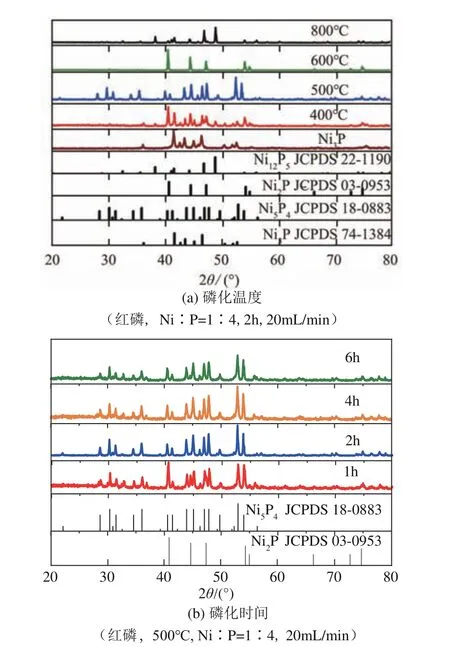
图1 不同磷化温度和磷化时间下制备样品的XRD谱图
根据文献可知,NiP和NiP分别可以在600℃和800℃的惰性气氛条件下稳定存在。因此,本文只选取NiP晶相考察500℃磷化温度下NiP晶相的稳定性,结果如图1(b)所示。磷化1h后,样品中NiP转变为NiP与NiP的混相,这与400℃磷化温度的影响一致,说明此时尚未达到晶相稳定的条件。磷化2h 后,样品中NiP 晶相消失,转变为NiP晶相;继续延长磷化时间至4h 和6h,样品中NiP晶相保持不变。这进一步表明磷化过程是一个晶相渐变的过程,在特定的磷化温度下可以形成稳定的晶相,且该稳定晶相不随磷化时间的延长发生进一步变化。
2.1.2 Ni/P摩尔比和磷化气速的影响
以红磷为磷源,在磷化温度为500℃、磷化时间2h 的条件下,使用不同Ni/P 摩尔比对其磷化的结果见图2(a)。当Ni/P 摩尔比为1∶1 时,样品以NiP晶相为主,仅有少量NiP晶相存在于样品中;逐渐增大Ni/P摩尔比到1∶4,NiP的特征峰消失,得到NiP晶相。继续增加红磷用量,当Ni/P 摩尔比为1∶6 时,在2=32.7°、55.7°处样品谱图中出现NiP(JCPDS 21-0590)的衍射峰,表明部分NiP进一步向NiP转变,形成NiP与NiP的混合晶相。随着红磷用量增加,NiP衍射峰强度提升,但即使Ni/P 摩尔比达到1∶12,仍无法合成出NiP纯相。可见随Ni/P摩尔比的改变,磷化过程中NiP样品会依次发生NiP→NiP→NiP的晶相转变现象,增加磷源用量有助于含磷量更高的NiP晶相生成,但无法获得纯NiP晶相。
除Ni/P 摩尔比外,磷化气速也会影响制备结果。如图2(b)所示,当气速为20mL/min时,样品为NiP晶相。降低气速至10mL/min,样品中出现NiP晶相,与低Ni/P 摩尔比下得到的晶相结果相似,部分NiP向NiP晶相转化。相反,增加气速至40mL/min,样品表现为NiP 与NiP 的混相,几乎没有NiP晶相出现,与高Ni/P 摩尔比下晶相结果相近。可见,磷化气速对晶相结果的影响近似于Ni/P摩尔比的作用,低气速下,磷源与NiP接触时间延长,提升了磷源浓度,促使Ni/P 摩尔比更高的磷化镍晶相生成。

图2 不同Ni/P摩尔比和磷化气速下制备样品的XRD谱图
2.1.3 磷源种类的影响
使用次亚磷酸钠代替红磷对NiP 进行磷化处理,以考察磷源种类对NiP晶相的影响。结果发现,与红磷相比,使用次亚磷酸钠作磷源时,仅能得到NiP(≤600℃)和NiP(800℃)晶相,未发现NiP晶相[图3(a)]。在磷化温度为500℃时,增加磷源用量[图3(b)]、调控磷化时间[图3(c)]或降低气速[图3(d)],磷化得到的样品仍为NiP晶相。可见,以次亚磷酸钠为磷源无法制备出NiP晶相,这说明单质磷是制备NiP的重要因素。
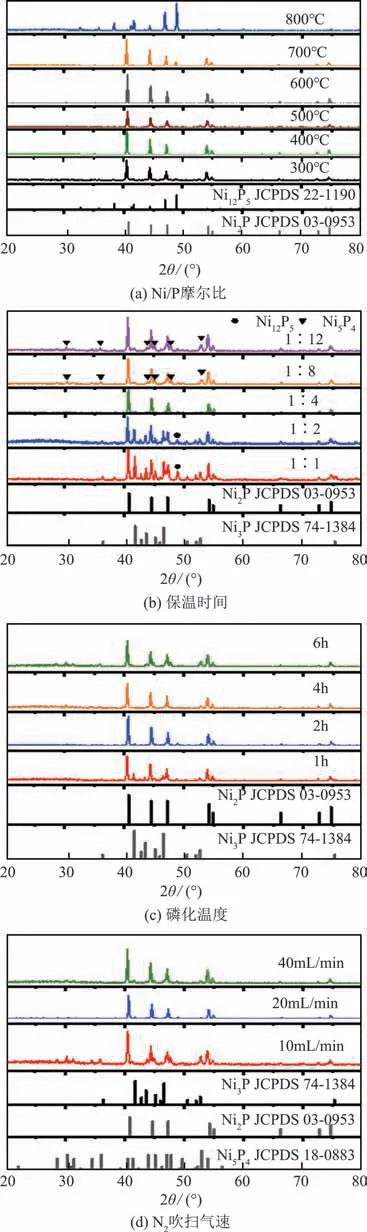
图3 使用次亚磷酸钠作磷源时,不同条件下制备样品的XRD谱图
2.2 NixPy的XPS分析
利用X 射线光电子能谱(XPS)对合成的4 种NiP晶相的表面组成和电子结构进行表征,结果如图4 所示。由图4(a)可知,NiP、NiP、NiP、NiP 样品中分别位于853.2eV、853.0eV、852.9eV及852.7eV 的谱峰归属为带正电的镍物种(Ni),电荷大小顺序为NiP>NiP>NiP>NiP。856.9eV、855.5eV、855.2eV 和855.0eV 的谱峰分别归属于NiP表面被氧化后Ni的谱峰。位于861.6eV、861.4eV、860.5eV 的谱峰归属为NiO 2p 轨道卫星峰。在图4(b)P 2p XPS 谱图中,位于129.2eV、129.3eV、129.5eV 及129.7eV 的谱峰分别归属于NiP、NiP、NiP、NiP 中 带 负 电 的 磷 物 种(P),且有130.1eV、130.2eV、130.3eV、130.6eV的谱峰依次对应NiP、NiP、NiP、NiP 的P 2p轨道峰,同样证实带负电荷的磷物种(P)的存在。此外,位于133.1eV、133.3eV、134.4eV、134.5eV的谱峰是钝化过程中在NiP表面生成的氧化态的磷物种。

图4 Ni5P4、Ni2P、Ni12P5、Ni3P的XPS谱图
2.3 NixPy/CdS光解水制氢性能
2.3.1 NiP/CdS的XRD结果分析
CdS 和质量分数5%NiP/CdS 复合材料的XRD图谱如图5 所示。CdS 样品的XRD 衍射峰在2=24.8°、26.5°、28.2°、36.6°、43.6°、47.8°、50.9°、51.8°、52.8°和58.3°处出现,与六方晶型CdS(JCPDS 41-1049)一致。NiP/CdS 复合材料中,CdS的衍射峰未发生偏移,说明NiP并未对CdS结构造成影响。除CdS衍射峰外,复合材料中可观测到归属于NiP晶相的特征衍射峰,但由于NiP含量较低,其衍射峰强相对较弱。该结果表明成功制备出NiP/CdS复合材料。

图5 CdS和5%NixPy/CdS复合材料的XRD谱图
2.3.2 UV-vis及PL结果分析
CdS 和4 种5% NiP/CdS 复合材料吸光性能由UV-vis 漫反射光谱测得,结果如图6(a)所示。CdS的吸收边约为540nm,对应带隙能约为2.3eV。任一晶相NiP与CdS 复合后,得到的NiP/CdS 样品吸收边与CdS吸收边一致,说明加入NiP并未改变复合材料的禁带宽度。复合材料在可见光区吸收强度有所提高,说明NiP助剂增加了样品在可见光范围内的吸光能力。但富金属的磷化镍的物理性质更接近于金属而非半导体,这意味着通过添加磷化镍助剂增强的吸收光不能转化为反应所需的光生电子和空穴对,也就是说磷化镍不能通过产生反应所需的光生电子-空穴对来提升复合材料的可见光利用率。
图6(b) 是CdS 和4 种5% NiP/CdS 复合材料的PL光谱。在使用相同波长的激发光对样品激发时,CdS相较其他样品载流子复合发光强度最高,说明CdS 中有严重的载流子复合现象发生。CdS 与NiP复合后,峰强度明显降低,且4种NiP/CdS复合材料峰强度依次为NiP/CdS< NiP/CdS< NiP/CdS
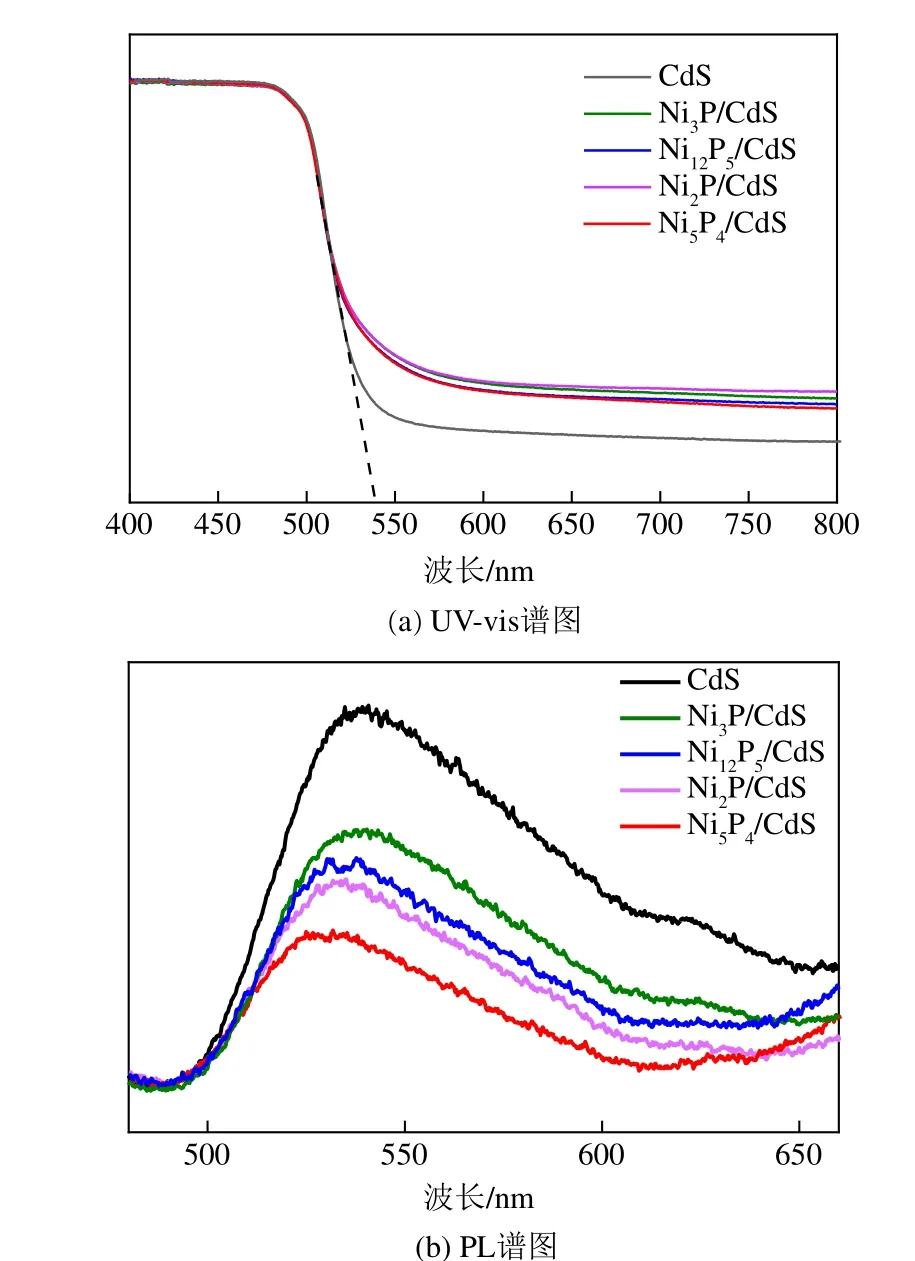
图6 CdS和5%NixPy/CdS复合材料的UV-vis谱图和PL谱图
2.3.3 NiP/CdS光解水制氢反应活性
对制备的CdS和4种5%NiP/CdS复合材料进行光解水反应性能评价,结果如图7(a)所示。CdS光催化活性相对较弱,制氢速率仅93.7μmol/(g·h),表观光量子转化效率为0.07%。当CdS 分别与4 种NiP助剂复合后,NiP/CdS 样品光催化活性显著提高,其中,以NiP、NiP、NiP、NiP为助剂的样品制氢速率依次可达306.5μmol/(g·h)、415.3μmol/(g·h)、502.1μmol/(g·h)和601.5μmol/(g·h),表观光量子转化效率依次为0.23%、0.32%、0.37%、0.46%。可见以磷化法制备的4种NiP晶相均可作为良好光催化助剂,显著提升CdS的光催化活性。NiP/CdS复合材料表现出最好的光解水活性,相较CdS产氢速率提升5.4倍。为评价NiP/CdS复合材料的反应稳定性,对NiP/CdS催化剂进行9次循环实验,每次循环前使用Ar置换管内气体,确保没有氢气残余,反应结果如图7(b)所示。在上述循环实验中,随循环次数增加,产氢量基本保持一致,可认为样品具备良好的稳定性。
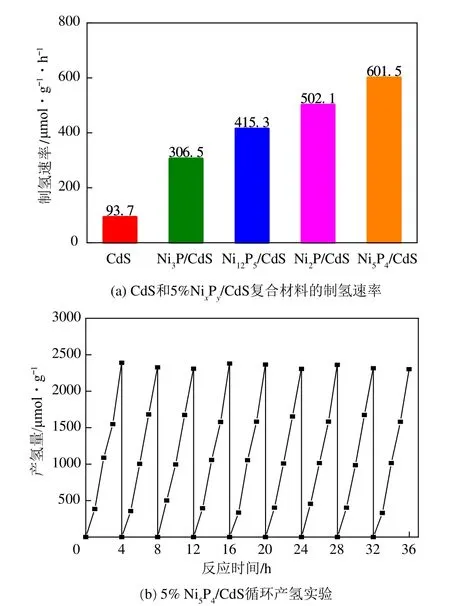
图7 CdS和5%NixPy/CdS复合材料的制氢速率和5%Ni5P4/CdS循环产氢实验
2.4 反应机理分析
对4类NiP/CdS复合材料光解水性能提升的机理进行分析,如图8所示,当NiP/CdS复合材料受到可见光照射后,半导体CdS 吸收光子带来的能量,产生电子跃迁,电子从价带被激发到导带,价带上留下带正电的空穴,从而产生一组光生电子-空穴对。之后得益于NiP较高的功函数,光生电子受NiP吸引,发生从CdS 到NiP的电子转移,在助剂表面发生HO 的产氢还原半反应。同时,CdS 价带上的光生空穴被牺牲剂NaS、NaSO捕获消耗。循环过程中发生的反应可以用式(2)~式(4)表示。

图8 NixPy/CdS复合材料光解水制氢机理示意图

反应过程中,由于NiP助剂的引入,光生电子在CdS上停留时间减少,从而载流子复合现象被抑制,材料的光解水制氢活性得到显著提升。
3 结论
本文以红磷为磷源,以NiP为前体,通过改变磷化温度、磷化时间、Ni/P摩尔比和磷化气速制备三种NiP(NiP、NiP、NiP)晶相。磷源种类和磷化温度是影响晶相的最主要因素。以红磷为磷源,当Ni∶P摩尔比取1∶4、N气速为20mL/min时,分别在500℃、600℃和800℃的温度下对NiP 磷化2h,依次制得NiP、NiP、NiP晶相。以次磷酸盐为磷源时,无法得到NiP晶相,当温度低于和高于800℃时分别得到NiP和NiP晶相。将上述4种NiP(NiP、NiP、NiP和NiP)作为助剂与CdS 制备光催化复合材料,得到的4 种NiP/CdS 复合材料均表现出良好的光解水制氢性能。其中,以NiP/CdS 效果最佳,制氢速率较CdS 提高5.4 倍,且三次循环实验中材料保持良好的稳定性。
