新型电活性离子交换材料MIL-101/Py复合机理的理论计算
2022-05-24延彩萍安小伟郝晓刚
延彩萍,安小伟,杜 晓,郝晓刚
(太原理工大学 a.化学工程与技术学院,b.环境科学与工程学院,太原 030024)
离子分离涉及盐湖资源利用、有色金属冶炼、生物医药开发及重金属污水处理等诸多领域,是化工和材料领域的研究热点。由于离子分离过程中,通常存在多种共存离子的干扰,因此如何实现高选择性分离是离子分离领域的主要挑战。目前,液相中离子分离的方法主要有沉淀法、吸附法、离子交换法、萃取法、电絮凝法、膜分离法和微生物法等[1-3]。上述方法利用目标离子和干扰离子之间的理化特性差异,结合不同的分离机制,能够有效地实现离子的选择性分离。但是当面临盐湖提锂、稀土分离、放射性废液处置等特殊工况时,由于待分离目标离子浓度极低、共存离子种类繁杂且干扰能力极强,现有的分离技术通常难以兼顾分离效果和分离成本。
电控离子交换(electrochemically switched ion exchange,ESIX)技术是在电吸附基础上,结合对特定目标离子具有选择识别功能的电活性离子交换材料(electroactive ion exchange materials,EIXMs),而开发的一种新型离子分离方法。将EIXMs制备成纳米尺度的薄膜,通过调控施加在膜上的氧化还原电位可以控制目标离子的置入与释放,该方法具有低浓度去除、速率可控、无二次污染、高选择性等优点[4]。EIXMs需兼具电子、离子双重传递性能,目前已经发现满足ESIX性能的材料主要包括两大类:含变价金属的无机电活性材料和导电高分子材料[5]。
以铁氰化镍(NiHCF)、锰酸锂(LiMnO4)、卤氧铋(BiOX)等为代表的含变价金属无机电活性材料,能利用其独有的晶格结构对特定目标离子进行筛分从而产生较高的选择性。研究表明,NiHCF对Cs+离子[6]、LiMnO4对Li+离子[7]以及BiOBr对Br-离子[8]分别表现出优良的选择性。而以聚吡咯(PPy)、聚苯胺(PANI)、聚乙烯二氧噻吩(PEDOT)等为代表的导电高分子能够凭借聚合过程中掺杂离子的印迹作用,实现对掺杂离子的选择性分离。LUO et al制备出一种电活性碘离子阱聚吡咯膜(PPy/I-),在I-和Br-质量浓度均为5 mg/L的混合溶液中I-/Br-的分离因子高达22.16[9].就分离性能而言,含变价金属的无机电活性材料晶格结构较为稳定,且尺寸均匀,因此选择性高,稳定性好,但已发现的材料种类有限,难以满足对诸多不同目标离子的分离需求。通过离子印迹制备的导电高分子,虽然适用性强,能够以不同离子作为印迹模板实现离子分离,但在离子置入/释放过程中会破坏高分子链的空间结构,从而显著降低其稳定性。因此开发更为高效、普适的新型EIXMs合成策略是该研究领域的重要方向之一。
金属有机骨架(metal organic frameworks,MOFs)作为一类种类多样、孔径均匀可调、孔道易于官能团化的新型材料,近年来备受关注。但由于大多数MOFs本身不具备电子、离子双重传导性能,无法直接作为EIXMs用于离子分离。为将MOFs多样、可调的孔道结构应用于ESIX领域,研究人员提出一种将导电高分子穿插在MOFs孔道内部,制备新型EIXMs的合成策略。巧妙地将聚合物单体作为客体分子引入MOFs的纳米孔道内,并通过原位聚合(穿插生长)生成高分子,利用导电高分子与MOFs原始孔道组合重构形成新的离子传递通道,同时将导电高分子作为电子传递通道,制备出一类新型的EIXMs.通过这一策略,研究人员成功合成了PEDOT/MIL-101[10]、PPy/UiO-66[11-12]等杂化材料。
调控导电高分子/MOFs杂化材料的孔道尺寸是实现电控离子选择性分离的关键,而这一过程取决于MOFs的原始孔径以及导电高分子在孔内的聚合量。导电高分子的聚合涉及单体之间及单体与MOF之间的相互作用,这种相互作用对高分子的聚合过程及导电高分子在孔道内的分布具有重要影响。因此,探索导电高分子单体在MOFs限域孔道内的传递扩散及聚合规律,对于控制离子传递通道的孔径及结构具有重要意义。针对这一问题,本文以PPy/MIL-101为研究体系,结合实验与理论模拟,从微观层面探索了吡咯单体在MOF孔道中的运动及吸附位点。MIL-101包含2.9 nm和3.4 nm两种类型的介孔笼[13],宽阔的孔道在提供高效的离子传输通道的同时可以容纳足够多的客体分子;吡咯单体大小约为0.4 nm,尺寸远小于MIL-101的孔窗,且具有易氧化、导电性好的优点[14]。MIL-101吸附吡咯单体达到上限后,利用吡咯单体在常温下易挥发的特性,通过在室温下干燥不同时间来控制MIL-101中留存吡咯单体的量。采用简单有效的方法巧妙地设计不同吡咯含量的MIL-101/Py电活性材料。通过不同含量调控孔径,为基于孔径筛分效应的离子分离奠定基础。理论计算用分子动力学和量化计算的方法,探究了不同含量吡咯单体在MIL-101中的分布规律及吡咯与MIL-101之间存在的相互作用。
1 实验及模拟计算
1.1 实验部分
将制备的MIL-101粉末称取0.2 g加入吡咯溶液中,超声使溶液分散均匀,室温浸泡3 d,使吡咯单体充分进入MIL-101的纳米孔中,抽滤得到MIL-101/Py复合物。室温下,将抽滤得到的MIL-101/Py用精密天平称量,且每30 s记录其质量。复合材料中吡咯绝对含量X(g Py/g MIL-101)由公式(1)计算:
(1)
式中:Gt为t(min)时湿物料的质量,g;Gc为干物料质量,即MIL-101的初始质量,g.
复合材料的干燥速率U(g·m-2·min-1)由公式(2)计算:
(2)
式中:ΔX是吡咯含量的差值;Δt是干燥时间间隔,min;S是复合材料的干燥面积,m2.
1.2 模拟计算模型及细节
1.2.1MIL-101计算模型
MIL-101晶胞属于Fd-3m空间群,为立方晶系,晶格常数a=8.886 9 nm,包含14 416个原子。MIL-101最基本的单元由Cr3O三核铬簇和对苯二甲酸构成,每个Cr3O簇中包含一个μ3O,4个羧酸氧和1个F或OH末端位点。为了减少计算量,在分子模拟中利用对称性将单元晶胞转化为原胞进行计算(图1(a)),将原胞进行几何优化以达到能量最小[15]。如图1(b),(c)所示,在量化计算中构建簇模型1和2用于研究MIL-101中Cr3O簇和有机配体与吡咯间的相互作用[16]。
1.2.2力场与电荷
本文所涉及的理论计算均使用Material Studio 8.0(MS 8.0)软件包完成。其中,分子动力学模拟采用Forcite模块进行,蒙特卡罗模拟采用Sorption模块完成。力场是经典分子力学模拟中的重要参数,通过匹配力场类型,可以分配各种势能项的参数,最终形成一个完整的势能面,用于计算原子之间的力。本文采用Universal力场(UFF)赋予MIL-101中每个原子力场,用Lennard-Jones势能模型描述体系中的非键作用[17],用Ewald求和方法描述静电力。
由于MS 8.0软件中的QEq方法不能用于计算全部原子的电荷,我们参考文献中的方法通过构建Cr3O簇模型来计算原子电荷[18-19]。采用Dmol3模块对模型进行几何优化,广义梯度近似(generalized gradient approximation,GGA)的 Perdew-Burke-Ernzerho(PBE)函数作为交换相关势,基组采用双数值加极化函数(double numerical basis plus polarization function,DNP).优化过程的能量收敛限度为1.0×10-5Ha、力的收敛限度为2×10-2Ha/nm、位移收敛限度为5×10-4nm[16].通过布局分析得到的静电势(ESP)电荷用于拟合原子电荷[20]。

图1 MIL-101的模型Fig.1 Model of MIL-101
在分子动力学模拟中,设置生产步数为106,平衡步数为105,对填充有不同吡咯单体的MIL-101/Py模型进行几何优化使整体能量最小化。执行动力学任务时选择NVT系综,并用Nosé算法调控受控温度下系统和环境之间的热量交换。

表1 MIL-101的LJ势能参数及原子电荷Table 1 LJ potential energy parameters and atomic charges of MIL-101
2 结果与讨论
2.1 干燥实验
MIL-101/Py在室温下的干燥曲线如图2(a)所示,MIL-101/Py中吡咯单体的挥发可分为3个阶段[10]。在0~15 min内(阶段Ⅰ),MIL-101/Py的吡咯含量下降最快,此时MIL-101/Py表面比较湿润,附着有大量的吡咯溶液,外层吡咯单体和MIL-101之间没有相互作用力,所以这段时间吡咯单体挥发最快;在15~55 min内(阶段Ⅱ),MIL-101/Py的吡咯含量下降速度减慢,经过第Ⅰ阶段的干燥后,剩余的吡咯单体与MIL-101内外表面的原子或有机配体之间存在分子间作用力,因此挥发速率比上一个阶段慢;在55~120 min之间(阶段Ⅲ),吡咯的挥发速度基本平稳,表明MIL-101中的吡咯含量趋于稳定,这对应于吸附在MIL-101纳米孔中的吡咯单体的挥发,这部分吡咯挥发受到吡咯分子间作用力及MIL-101骨架原子与吡咯之间作用力的阻碍。由图2(a)求微分得到干燥速率曲线如图2(b)所示,可以看出Ⅰ、Ⅱ阶段均为物料的减速干燥阶段,当吡咯含量为0.113 2 g/g MIL-101时,干燥速率达到恒定值。

图2 室温下MIL-101/Py中Py的干燥曲线(a) 及干燥速率曲线(b)Fig.2 Drying curve (a) and drying rate curve (b) of Py in MIL-101/Py at room temperature
2.2 MIL-101/Py复合模型及动力学研究
针对上述实验结果,采用蒙特卡罗方法构建对应干燥实验不同阶段的MIL-101/Py模型,探究吡咯进入MIL-101的过程以及它们之间的相互作用。从图3可以看出,约723个吡咯单体被吸附到原胞
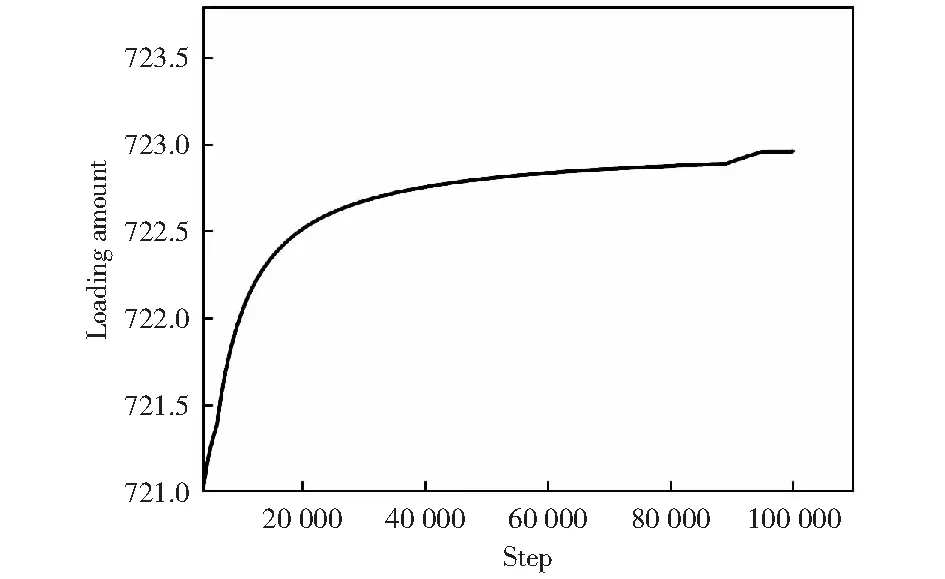
图3 常压下MIL-101原胞吸附吡咯单体的平衡曲线Fig.3 Equilibrium curve of adsorption of pyrrole monomer by MIL-101 primitive cell under normal pressure

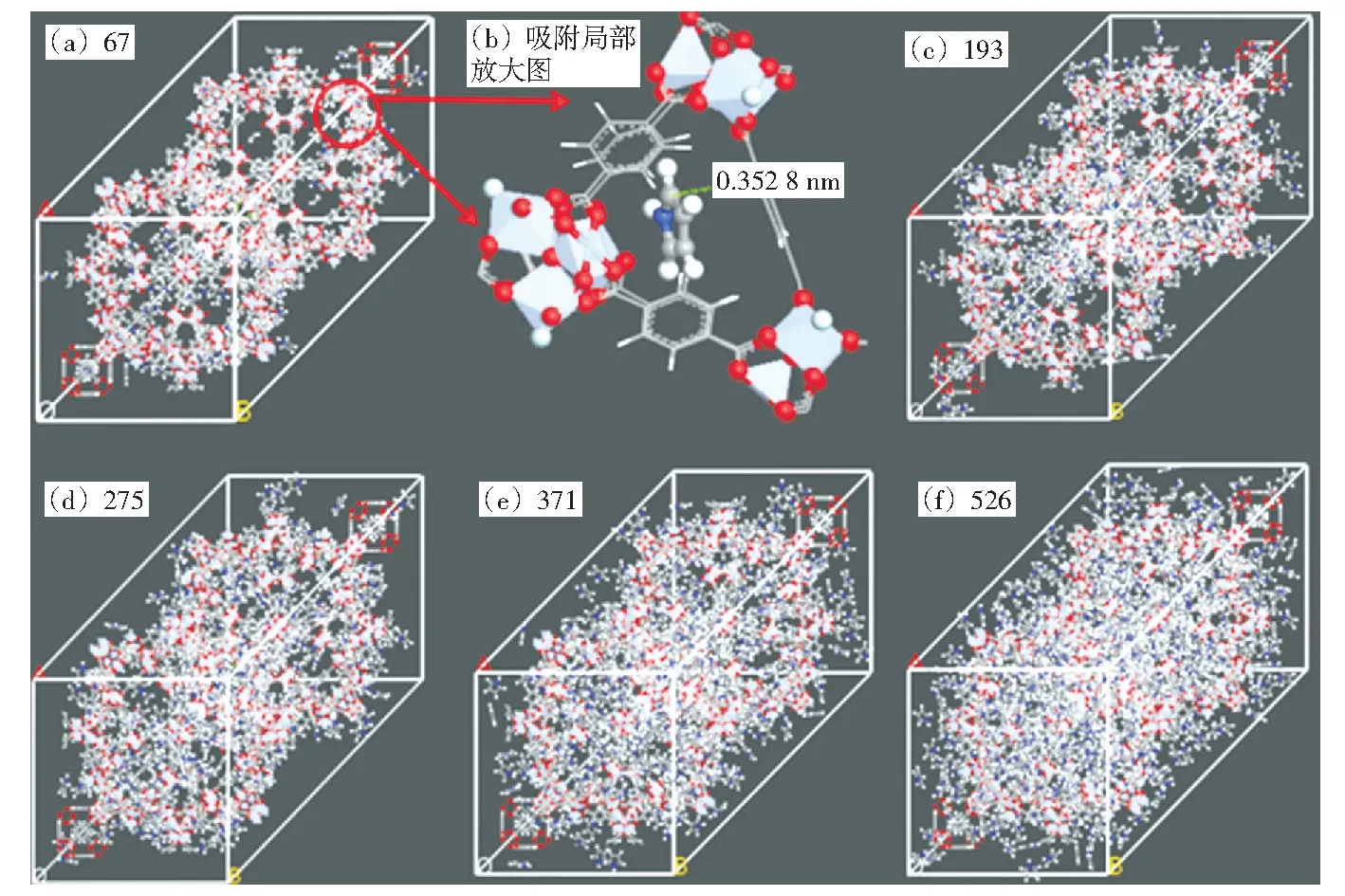
图4 MIL-101吸附不同数量吡咯单体的构型图Fig.4 Configuration snapshots of MIL-101 adsorbing different amounts of pyrrole monomers
不像IRMOF-1(Zn)和IRMOF-8(Zn)孔形状为简单的立方形[21],MIL-101的孔道更复杂,难以通过密度分布图直接看出吸附质在孔道中的分布。而相对浓度分析可以为吡咯在MIL-101中的分布规律进行进一步说明。图5为绝对吡咯含量为0.53 g Py/g MIL-101时,对应模型进行500 ps动力学模拟获得的相对浓度分布。图5(a)表明,当t=0 ps时,以晶胞某一处为原点,在不同距离处吡咯相对浓度范围波动不大,说明MIL-101中各处的浓度基本恒定;而500 ps后,曲线在12 nm~35 nm范围内呈现下凹趋势,对应的是介孔笼的中心部分,这表明笼中心吡咯单体的相对浓度远低于边缘位置。同时,分析MIL-101晶胞不同晶面的相对浓度,出现相似的趋势,证明当吡咯负载量较低时,吡咯单体确实优先分布在MIL-101笼的边缘(图5(b)).

图5 (a)不同吸附时间吡咯在MIL-101中及(b) t=500 ps时 吡咯在MIL-101不同晶面的相对浓度分布Fig.5 Relative concentration distribution of pyrrole in MIL-101 at different times (a) and at different crystal planes of t=500 ps (b)
2.3 吸附位点
吸附能和原子间距离是研究吸附位点的重要标准,可以确定吸附方式和吸附强度。吸附能的绝对值越大说明二者的相互作用越强;而吸附位点之间的距离对于化学键和氢键的形成,是一个重要的参数[22]。通过比较吡咯单体在MIL-101骨架不同原子位置优化前后的结构及结合能,定量研究MIL-101吸附吡咯的优先位点。将吡咯沿N和C原子的方向分别放置在Cr3O簇的F、Cr和O原子周围,分别记为吸附N位点和C位点。比较两个位点的结合能发现,与C位点相比,整体上簇模型与吡咯N位点的结合能更大,说明簇模型与吡咯的N位点相互作用更强。
对N位点吸附进行进一步分析。在Cr3O-Cr体系中(图6(a),(d)),几何优化后吡咯靠近O原子,且与O结合能更大,这是因为MIL-101与吡咯的相互作用源于静电力,而静电力强度弱于Cr3O-O体系形成的氢键。由图6(b),(e)发现,几何优化后H与F原子间距离dH—F=0.161 3 nm,N—H—F之间的角度θ=175.87°,与文献中N—H…F体系的键长dH—F=0.163 nm、键角θ=179°一致[23],证明Py与F原子之间N—H…F分子间氢键的形成。图6(c),(f)为Py与O原子相互作用几何优化前后的结构示意图,同样形成了分子间氢键,其键长dH—O=0.214 9 nm,键角θ=150.56°,与经典N—H…O体系有一定的差异[24],是体系不同、原子间协同作用不同而引起的。表2表明N—H…O体系吸附能大于N—H…F,是因为虽然F元素电负性大于O,但不同于F-,化合物中的F因为极化率低而易导致弱氢键的形成[25]。此外,氢键的大小除受原子电负性影响外,还受静电作用、空间位阻、π共轭及体系协同作用的影响[26]。图7的电子密度图中,蓝色和红色区域分别代表富电子和缺电子,相比于N—H…F,N—H…O重叠部分更明显,表明在O与N—H之间存在更强的电子转移,相互作用更强,与上述结论一致。

图6 Cr3O簇模型与吡咯的N位点几何优化前(a,b,c)、后(d,e,f)的结构对比Fig.6 Structure comparison of Cr3O clusters and the N sites of pyrrole before (a,b,c) and after (d,e,f) geometry optimization

图7 N—H…F(a)和N—H…O(b)体系的电子密度图Fig.7 Electron density maps of N—H…F(a) and N—H…O(b) systems
在探索吡咯与MIL-101有机配体的相互作用时,为了降低F、O、Cr对吡咯的影响,采用模型2进行量化计算。二者相对位置垂直时,结合能为-55.296 kJ/mol,相对位置平行时,结合能为-30.765 kJ/mol,与CH-π氢键的能量相近[27]。用于定义CH-π氢键的Brandl-Weiss系统体系表明供体与受体之间满足特定的几何标准,即供体碳与芳族体系环质心之间的距离(dC—M≤0.45 nm)[28].图8为吡咯与苯环相对位置平行时,几何优化前后的模型对比图。图8(c)中最小距离为dN-C6=0.365 8 nm,最大距离为dN-C3=0.400 1 nm<0.45 nm,而N原子的投影在苯环中心,可以推测出dC-M<0.45 nm,符合CH-π体系的标准。由此可以证明体系中CH-π/NH-π键的存在,且CH-π是一种相对较弱的、非规范的氢键。
综上所述,吡咯与MIL-101之间相互作用包括:1) 吡咯与Cr3O中Cr的静电相互作用,与F、O间的N—H…F和N—H…O的经典氢键作用;2) 吡咯与有机配体苯环的CH-π/NH-π氢键作用。计算得到MIL-101中各部分对吡咯吸附的强弱顺序为:Cr3O-O>Cr3O-F>CH-π/NH-π>Cr3O-Cr,且由吸附能判断,其中Cr3O-O体系吸附能最大为-91.081 kJ/mol(<1.6 eV=154.4 kJ/mol,物理吸附)[29]。所以,吡咯与MIL-101之间的吸附方式均为物理吸附。

图8 MIL-101的有机配体与吡咯几何优化前(a)、 后(b),(c)的结构对比(平行)Fig.8 Structure comparison of organic ligands of MIL-101 and pyrrole before (a) and after (b),(c) geometry optimization (parallel)

表2 吡咯与MIL-101在不同吸附位点的结合能Table 2 Binding energies of pyrrole and MIL-101 at different adsorption sites
3 结论
1) 采用浸渍法结合吡咯易挥发的特性,简单、有效地制备了不同吡咯含量的MIL-101/Py复合电活性材料。
2) 将实验对应的一系列模型进行分子动力学模拟,研究了MIL-101中吡咯单体的运动状态。发现吡咯倾向于分布在MIL-101介孔笼的边缘然后向中心聚集。吡咯单体在MIL-101腔体中原位聚合后,聚合物链在MOF中的限域效应会调整笼有效空间,为基于孔径筛分效应的离子选择性分离奠定基础。
3) 考察了吡咯与MIL-101的Cr3O簇及有机配体的相互作用。DFT理论计算表明,MIL-101吸附吡咯存在氢键及静电作用两种机理。计算得到MIL-101中各部分对吡咯的吸附强弱顺序为:Cr3O-O>Cr3O-F>CH-π/NH-π>Cr3O-Cr,吸附方式均为物理吸附。鉴于吡咯中吸附位点以N位点为主,理论计算可以为含氮类有机物的去除提供参考价值。
