吸烟相关性间质性肺疾病研究进展
2022-05-24罗干倪吉祥杨玉婷徐彪
罗干 倪吉祥 杨玉婷 徐彪
作者单位:443000 湖北 三峡,三峡大学人民医院/宜昌市第一人民医院呼吸与危重症医学科
长期以来,吸烟往往被认为与慢性阻塞性肺疾病及肺癌关系密切。但近年来,人们对烟草成分导致肺损伤机制的认识不断提升,伴随着对吸烟相关性间质性肺疾病的认识也在不断发展。在这类相关疾病(表 1)中呼吸性细支气管炎伴间质性肺疾病、脱屑性间质性肺炎、肺朗格汉斯细胞组织细胞增多症、急性嗜酸性粒细胞性肺炎的相关机制与吸入烟草有明确清晰的关联。但对吸烟相关性肺间质纤维化、肺纤维化合并肺气肿综合征以及结缔组织引起相关的间质性肺疾病的作用和机制尚不明确。在这篇综述中我们将重点关注吸烟介导肺损伤的机制以及这些疾病的临床表现、影像特征、病理特点及预后。

表1 与吸烟相关的间质性肺病
吸烟介导的肺部炎症和纤维化。
众所周知,香烟中含有多种有害化学物质(特别是尼古丁、煤焦油、一氧化碳),是导致呼吸道和肺部病理变化的重要危险因素[1]。香烟烟雾中的反应性氧化剂、尼古丁和其他毒素,诱导上皮细胞和内皮细胞活化,通常分泌多种炎症细胞因子和趋化因子,这些细胞因子和趋化因子诱导免疫细胞过度聚集进入各种肺组织,包括小气道和大气道以及肺实质[2]。根据现有研究表明,烟草烟雾在小气道病理过程中的作用已有详细记载[3]。尼古丁作为一种在香烟中发现的主要化学物质,它能促进肺泡上皮和血管内皮的损伤,还能诱导 TGF-β1 的产生和释放,也能增强炎症细胞(巨噬细胞、中性粒细胞)的聚集(图 1)和活性氧的产生,而且对成纤维细胞和肌成纤维细胞产生相关基质(包括胶原蛋白、纤连蛋白和弹性蛋白)至关重要[4-5]。由香烟烟雾其他成分直接诱导的血小板衍生生长因子(plateletderived growth factors,PDGF)能促使成纤维细胞和肌成纤维细胞增殖,因此它可通过增加产生基质蛋白的细胞数量来增强 TGF-β1的效应[6]。近年来对吸烟引起弥漫性肺部疾病患者的肺活检组织,进行免疫组化研究显示TGF-β1和PDGF 表达均上调,提示这些细胞因子在纤维化演变中起关键作用[7]。众所周知,端粒是执行染色体稳定性的特殊核苷酸重复序列。当人体进行正常的有丝分裂时,端粒长度随着细胞分裂而逐渐缩短,当达到相应的长度时,会激活持续性DNA损伤反应,进一步导致细胞凋亡[8]。p53是在衰老细胞中表达的一种抑癌基因,一般处于失活状态,当应激反应导致DNA损伤时,p53相应磷酸化激活周期性依赖蛋白激酶抑制因子p21的表达上调,继而触发细胞凋亡[9]。这一现象已在 IPF 的肺泡上皮细胞和外周血细胞中得到描述[7]。
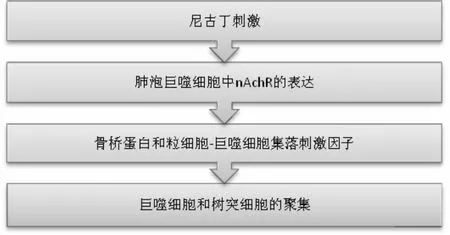
图1 尼古丁刺激导致炎症细胞的募集nAchR-烟碱乙酰胆碱受体
呼吸性细支气管炎-间质性肺炎
1974年Dennis Niewoehner[10]等人首次将呼吸性细支气管炎(respiratory bronchiolitis,RB)描述为因吸烟所诱发的特殊病理表现,他观察到这种病变是由远端气道及支气管周围肺泡中巨噬细胞(含有色素沉着)聚集所引起。呼吸性细支气管炎在吸烟者很常见,通常无临床症状。因其他原因接受肺活检的吸烟者中,达到89%的病例多为偶然发现[11]。肺功能绝大多数呈轻度通气功能障碍[12]。胸部CT仅仅表现为支气管周围间质纤维化。若影像学上纤维化病变特征异常明显,那么呼吸性细支气管炎就会伴有间质性肺疾病(respiratory bronchiolitis-interstitial lung disease,RB-ILD),这就可能导致临床症状的出现。RB-ILD 患者通常有超过30年的吸烟史。通常发生在30~60岁之间,男性患者居多。根据国际流行病学报告数据显示,在间质性肺炎病例中RB-ILB的发病率为2%~13%[11, 13]。与许多弥漫性肺部疾病一样,临床表现主要是非特异性的,包括慢性咳嗽和呼吸困难[13]。
RB-ILD病变范围相比之下更为广泛,病变位置可达肺间质及导致肺泡炎性浸润。组织病理学上,呼吸性细支气管和肺泡管内存在褐色色素巨噬细胞,与轻度支气管周围间质增厚有关,这种增厚是由成纤维细胞、胶原沉积和单核细胞浸润共同引起的[13]。常见的表现还包括细支气管及周围肺泡纤维化,这种纤维化,会拉伸相邻的肺泡间隔,导致小叶中央型肺气肿[13-14]。几乎所有临床表现为慢性病程,肺部听诊可为正常,也可听及吸气相呼吸音。有文献报道杵状指表现并不常见,但当并发杵状指后,需警惕肺癌发生的可能性[15]。尽管 RB-ILD 具有细支气管扩张的表现,但肺功能检查通常显示混合性通气功能障碍,但主要是限制性功能缺陷,一氧化碳弥散量(carbon monoxide diffusing capacity ,DLCO)轻度至中度减少[13]。然而,在患有弥散性血管内凝血的患者中,一氧化碳弥散功能可能会严重降低。因此,糖尿病周围神经病变患者可能会死亡,但在 ILD (interstitial lung disease)患者中没有相关死亡的描述[16]。RB-ILD典型的高分辨率CT(high resolution CT,HRCT)包括小叶中央型毛玻璃样结节、斑片状毛玻璃样阴影(ground-glass opacity,GGO),支气管壁增厚和毛玻璃样衰减区[13-14]。
目前RB-ILD 的预后多数较为良好,这种疾病通常有一个稳定的病程,无需特殊治疗,戒烟是首要且最重要的治疗干预方式。尽管可以延长疾病生存期,但无论当前的干预治疗效果如何,在长期的随访中都可以观察到临床预后和生理改善是延迟的。在 Portnoy[17]等人的长期随访研究中,75%的患者存活了 7年以上,但临床预后表现良好和生理功能改善的患者分别只有占28%和10.5%。小型回顾性观察研究显示,皮质类固醇治疗偶尔会改善症状,但只有在肺功能明显功能障碍的情况下才应考虑使用;尽管在使用皮质类固醇和其他免疫抑制剂进行了治疗后,但仍有一些患者的临床生理参数可能会恶化[13, 17]。在皮质类固醇治疗和戒烟后的HRCT随访中,显示43%的患者支气管壁增厚、小叶中央型结节和磨玻璃样病变程度较前明显下降[18]。
脱屑性间质性肺炎
脱屑性间质性肺炎(desquamativeinterstitial pneumonia,DIP)最初是由Averill Liebow[19]博士等人在 1965 年提出描述,他们认为DIP 是一种独特的间质性肺炎,其特征是上皮细胞脱屑和不同程度肺泡填充。现已认识到巨噬细胞对肺泡空间的均匀填充是弥散性肺损伤的组织学标志,低倍镜下观察,最主要的表现是弥漫性及均匀性地肺部受累[20]。90%的DIP患者与吸烟有关,在极少数情况下,与其他外部疾病、自身免疫性疾病、感染或药物摄入相关[11]。75%的患者有粉尘或烟雾暴露史,92%的病例记录了特定类型的颗粒暴露。这些发现表明DIP与暴露于某些颗粒物之间存在关联[11]。
脱屑性间质性肺炎通常好发于40~60岁男性,男女比例为2:1。报告病例中的吸烟率高达60%~87%[21],与RB-ILD一样,临床表现为非特异性,如咳嗽、活动后气喘等症状,但与之不同的是,超过50%的患者出现杵状指。体检发现 60%的患者肺部出现吸气性啰音。限制性通气功能障碍和DLCO减少是判断疾病严重程度的重要指标[22]。组织病理学上,弥漫在 DIP患者肺泡中的大量巨噬细胞非常类似于 RB-ILD中出现的巨噬细胞,然而,DIP 可以在组织学上与 RB 相区别[20]。DIP组织病理学的主要特征是肺泡腔被巨噬细胞弥漫性填充,肺泡间隔弥漫性增宽,表现为纤维化非特异性间质性肺炎(non-specific interstitial Pneumonia,NSIP)模式。这些巨噬细胞与肺泡内的肺腺泡一起广泛分布,通常含有不同程度的浅棕色色素,巨噬细胞胞浆内可见细粒铁颗粒,多核巨细胞内可见嗜酸性粒细胞和淋巴样聚集物[20-22]。相比之下,脱屑性间质性肺炎往往具有更大程度的淋巴滤泡和嗜酸性粒细胞浸润。在常见的间质性肺炎(usual interstitial pneumonia,UIP)中发现的成纤维细胞病灶、蜂窝状是脱屑性间质性肺炎中缺乏的显著特征[21]。DIP 患者胸部 CT 表现为双肺散在的磨玻璃影,内部可见多发的薄壁囊状影。在下叶分布更明显,由于缺乏细支气管中央型分布,通常形成马赛克状衰减[21,23]。
研究表明DIP的发展终向比IPF好,10年生存率>70%,尽管它的预后可能比 RB-ILD 差,并且可能随着持续性吸烟而进展[21]。主动和被动避免接触烟草是治疗的基础。戒烟可使大约 20%至 50%的患者获得临床改善。在患者随访期间,非吸烟者的平均生存期为8.8年,吸烟者的平均生存期为7年[21]。全身性皮质类固醇给药的方式通常会维持病情稳定,但是很少改善症状。在长期治疗的情况下,可以考虑与环磷酰胺或硫唑嘌呤免疫抑制剂联合使用来减少类固醇的消耗[21-22]。然而,总体而言,关于 DIP 药物治疗效果的证据很少,大多数基于回顾性观察研究和病例报告。如果满足所有筛选标准,肺移植可能是一种选择[24],肺移植可使生存时间平均延迟5年[7]。
肺朗格汉斯细胞组织细胞增多症
肺朗格汉斯细胞组织细胞增多症(pulmonary Langerhans cell histiocytosis,PLCH)是一种以肺中CD1树突细胞多克隆积聚为特征的疾病。好发于20至40岁的年轻人,男女患病比率无明显差异。吸烟是成年人患这种疾病的最大危险因素。据报道,至少有90%的PLCH患者有吸烟史,更重要的是这种疾病会影响到二手烟接触史的人[25]。虽然疾病的确切机制尚不清楚,但很可能 PLCH 代表了一种髓样肿瘤,在大多数情况下表现为由吸烟引起的炎症特性。虽然提出的 PLCH 发病机制是涉及淋巴细胞和免疫细胞的免疫反应,但激活细胞增殖的吸入性抗原似乎存在于烟草烟雾中[25-26]。在小气道持续存在的肺郎格汉斯细胞(Langerhans cells,LCs)很可能是第一个致病因素[27]。吸烟可通过多种途径实现这一点。香烟烟雾促进呼吸道上皮细胞产生相应细胞因子。还能促进呼吸道黏膜对吸入性抗原的反应,来增强树突状细胞对感染因子和过敏原的激活. 还通过诱导细胞反应蛋白促进树突状细胞的存活[27-28]。现在已经揭示了吸烟对影响细胞周期途径突变的识别这条通路,包括 BRAF-V600E 突变(在高达 50%的 PLCH 病例中被识别)和丝裂原活化蛋白激酶 (MAPK),这可能解释了在某些个体戒烟后仍可观察到疾病进展[28]。
咳嗽和呼吸困难是最常见的表现特征,尽管高达四分之一的患者可能无症状.但三分之一的患者可能有全身性体质症状,如体重减轻、发热、盗汗和厌食,这为进一步检查以诊断隐匿性恶性肿瘤提供了线索。PLCH和其他恶性肿瘤之间的联系已经得到相关描述,其发生率从儿童的2.6%到成人的32%之间[26]。在一项大型队列研究中,单系统疾病和多系统疾病分别占患者的50%。在多系统疾病患者中,约15%的患者涉及危险器官[29]。约 15%的患者出现自发性气胸,其中一些是复发性的。患有胸部以外疾病可能会出现与皮肤、淋巴结、垂体或骨质受累有关的症状[30-31]。骨骼是最常见的受累系统,大约80%的PLCH患者存在骨骼病变[26]。除了肺实质受累外,肺血管受累导致的肺动脉高压在晚期原发性肺动脉高压患者中似乎相对常见。肺功能检测在疾病早期可为正常,也可表现出阻塞性、限制性、混合性通气功能异常。大多数患者的胸片通常是非特异性,表现双侧小结节或网状结节浸润。在晚期疾病中,以囊性改变和过度膨胀为主[14, 30]。当结合吸烟史和HRCT扫描的特征性影像学改变,可以对 PLCH进行临时诊断[24]。虽然确诊需要肺活检,但对于无症状或症状轻微的患者,尤其考虑治疗方式是戒烟的患者,肺活检并不是强制性的。在需要确诊的情况下支气管镜检查可能有助于诊断。在支气管肺泡灌洗液中检查出大于5%的 CD1a 阳性染色细胞(一种特异性的淋巴细胞标记物),可考虑 PLCH,但特异性相对不高。 PLCH的组织病理表现随着时间的推移而演变,因为该疾病存在于细胞性疾病到纤维化疾病的范围内。当朗格汉斯细胞被激活时,它们聚集在终末和呼吸细支气管周围。随着时间的推移,朗格汉斯细胞、嗜酸性粒细胞、浆细胞、淋巴细胞、多形核中性粒细胞和色素细胞的聚集在小气道周围,形成弥散的细小支气管中央型结节。光学显微镜显示肉芽肿样结节主要由朗格汉斯细胞组成,镜下表现为典型的苍白细胞质和肾形细胞核,其特征为CD1a 抗原或朗格林染色阳性,然而,伴随严重的肺动脉高压和弥散功能障碍可能会妨碍进行肺活检[7, 25-26, 28, 30]。
PLCH 预后相对较好,特别是对于能够戒烟的人。在诊断肺功能时测定结果显示正常或轻度减少的患者通常长期表现良好。回顾性分析显示经肺活检证实的疾病患者的 5 年生存率为 74.6%,10 年生存率为 63.9%.对于难治性或进行性 PLCH 患者,一些基于小样本的疗效证据,已经使用了其他疗法(皮质类固醇、免疫抑制剂和化疗)[28]。Grobost[32]等人还报告了一系列接受克拉屈滨治疗的进行性PLCH患者的改善结果(呼吸困难改善和一秒用力呼气容积增加)。在晚期疾病中,应考虑肺移植。即使仍有20%的复发机会,PLCH 患者在肺移植后的表现往往令人满意,移植后 10 年存活率几乎为 50%[28, 30]。
急性嗜酸性粒细胞性肺炎
急性嗜酸性粒细胞性肺炎(acute eosinophilic pneumonia,AEP)是一种急性呼吸系统疾病,表现为弥漫性肺浸润、低氧性呼吸衰竭、支气管镜下肺泡灌洗液嗜酸性粒细胞增多(支气管镜检查显示,在没有寄生虫感染、特应性反应或哮喘的情况下,嗜酸性粒细胞超过 25%)[31]。在许多情况下,AEP的病因仍不清楚。但几项研究提出吸烟可能与AEP发病机制相关。Hiroshi[33]等人通过对33名患者在AEP发作前的吸烟习惯做出了详细记录,并进行了相关烟雾激发试验,证明了吸烟是AEP发展的一个潜在病因,特别是吸烟习惯的改变。即使是短期被动吸烟也会导致AEP。大多数AEP患者年龄约30岁,男性为主,占60%。2/3 AEP患者目前都是吸烟者[34]。根据三项研究记录了[35]AEP患者的详细吸烟史,显示80%的患者在AEP发病前有不到两个月的吸烟史。此外,4%的患者在AEP发病前重新吸烟或增加吸烟量[36]。导致 AEP 病的原因还有很多,包括感染、药物反应和免疫性疾病[32]。嗜酸性粒细胞是骨髓来源的细胞,是免疫系统的重要组成部分。它们普遍暴露于外部环境中,可作为对寄生虫、真菌和其他生物体侵入的有效防御。除了直接损伤组织细胞能力之外,这些嗜酸性粒细胞颗粒的成分也可导致肥大细胞功能的上调、T 细胞增殖、血管通透性增加和平滑肌收缩.因此,尽管它们作为一种有效的防御手段,但嗜酸性粒细胞异常增殖和活化可导致广泛的组织损伤[31-32]。
在嗜酸性粒细胞引起组织损伤的同时,可伴随血液和蛋白质流出至组织间隙。组织学上,这一过程会引起弥漫性肺泡损伤及肺泡嗜酸性粒细胞明显浸润。急性AEP患者的肺组织病理学标本可见肺实质有密集的嗜酸性粒细胞浸润区,伴有弥漫性肺泡损伤的叠加改变。嗜酸性粒细胞可见于肺泡和间质内。其他特征包括嗜酸性脓肿、肺泡中有组织的纤维蛋白渗出物、2型肺泡细胞增生[11, 30- 31, 37]。胸部 CT 上的主要表现为双肺斑片状实性病变和磨玻璃样改变、大多数患者有小叶间隔增厚,约一半患者有胸腔积液的表现[7, 35]。诊断标准在不同来源之间有所不同,但通常包括急性发热性疾病、低氧性呼吸衰竭、胸片上弥漫性肺泡和/或间质性混合表现、嗜酸性粒细胞含量>25%、排除感染性病变[31, 36]。尽管支气管肺泡灌洗液显示嗜酸性粒细胞百分比较高,但外周血嗜酸性粒细胞计数通常是正常的.因此排除感染性病变很重要[38]。
AEP 患者对皮质醇有快速的疗效反应,大多数患者在 24 至 48 小时内有显著的临床改善。影像学表现及肺功能异常在一个月内得到改善。虽然皮质醇的最佳剂量目前尚不确定,但对于呼吸衰竭患者,静脉注射甲基泼尼松剂量为每6小时60至125毫克,直至症状改善后开始口服泼尼松[7, 35, 39]。回顾性研究表明,2%~67%的病例需要短期支持性通气,这取决于该疾病的风险组合性[35]。研究表明,短期疗程口服小剂量泼尼松足以治疗无呼吸衰竭的患者[39]。
与吸烟相关的其他临床病理综合征
一、吸烟相关性特发性肺间质纤维化
吸烟相关性特发性肺间质纤维化(smoking-related interstitial fibrosis,SRIF)是一种相对较为新颖的概念。主要在病理学家中获得了突出地位,尽管它可能在早期被描述为不同的术语,如“伴有纤维化的呼吸性细支气管炎”或“与纤维化相关的间质性肺病”[40]。Katzenstein[41]等人将SRIF 描述为慢性间质性纤维化的一种独特形式。根据病理学标本的各种小型研究显示,肺部 SRIF 相关变化的发生率在 14%至60%之间,但在重度吸烟者中发生率明显增加[41]。
吸烟相关性间质性纤维化的HRCT上最常见的表现为双侧小结节浸润和磨玻璃样改变。但在某些病例中,SRIF在HRCT 具有独特的影像学表现,包括上叶肺气肿和靠近胸膜下病灶,有时伴有网格状结构改变[37]。组织病理学上,SRIF表现为与肺气肿和RB相关的透明间质纤维化。增厚的肺泡间隔内可见致密的嗜酸性胶原沉积,通常伴有混合的增生性平滑肌束[41]。肥大的平滑肌束可伴随纤维化,但纤维化仅限于胸膜下和支气管周围间质。SRIF与UIP的透明嗜酸性胶原沉积表现不同,它可使肺泡间隔不同程度增厚,并扩大肺气肿和RB的间隙。肺泡间隔增厚在低倍镜下很容易发现,它往往分布于肺气肿相关的近胸膜下实质中,但也可以在其他实质中发现,包括小叶中心和与肺气肿无明显关联的随机区域[37, 41-42]。仅在少数患者中报道了肺功能测定,显示肺容量保持不变,1 秒内用力呼气量减少,并且一氧化碳弥散功能不成比例地相对性减少[42]。
鉴于公开研究的SRIF患者数量少,随访时间短,因此对SRIF的进展知之甚少。但目前所有研究都记录了随着时间推移,病程总体上趋于稳定,虽然一些患者可能表现出阻塞性通气障碍,但临床症状一般轻微,相对来说SRIF的自然发展病史可能比其他纤维化肺疾病更有利[7, 18]。在大多数患者中,SRIF是在因其他原因切除的肺组织中偶然发现的。Katzenstein[41]等人研究的9名患者,平均年龄65岁,目前都为吸烟者或曾经是吸烟者,吸烟者年限平均48年。五名男性和四名女性临床表现及病理变化没有出现性别差异,SRIF引起的特定症状也没有发生任何变化。9名患者中有6名肺功能检查发现轻至中度阻塞性通气功能障碍,4名患者出现弥散能力轻至中度下降。所有患者术后平均随访16个月均存活,无一例出现进行性呼吸障碍。
二、肺纤维化合并肺气肿综合征
肺气肿和吸烟之间的联系已被充分证实。然而,当肺气肿和肺纤维化在同一个患者中共存时,它代表了一种称为肺纤维化合并肺气肿的独特综合征(combined pulmonary fibrosis and emphysema,CPFE)。随着研究深入发现,CPFE 越来越被认为是吸烟者中的一个独特的实体,不再被认为是代表纤维化之上的肺气肿[43]。CPFE综合征在大量吸烟史男性患者中常见,吸烟史超过40包/年。中位生存期在2.1至8.5年之间,而5年生存率介于38%~55%[44],几乎是IPF的两倍,但与单纯肺气肿患者相比,总生存率更差[43]。在肺部CT发现肺纤维化和肺气肿改变的个体中,患病率从18.8%到50.9%不等,所有病例均为男性吸烟者[45]。
由于肺气肿的通气功能与肺容量和肺顺应性增加相关以及肺纤维化具有相反的生理效应,所以肺功能测试结果通常显示正常或一秒钟用力呼气容积(FEV1)/用力肺活量(FVC)相对正常,但肺对一氧化碳的弥散功能严重降低[43]。此外,CPFE 患者的肺动脉高压出现率更高,诊断时若出现肺动脉高压,则预示着总生存率降低。与单纯肺气肿或IPF患者相比,CPFE患者的肺癌发生率可能更高。1 项研究表明,CPFE 患者的肺癌发病率为 42%,而先前的研究表明,肺气肿患者的发病率约为 14%,肺纤维化患者的发病率约10%至15%(表 2)。对于CPFE而言,间质纤维化的组织病理学表现最常见于UIP,在少数报道的病例中为NSIP[16, 43]。胸部HRCT上CPFE的定义为上叶为主的小叶中央型肺气肿改变,下叶为主的弥漫性肺实质病变伴网格状改变,常表现为蜂窝样病变、牵拉性支气管扩张[42-44, 46]。
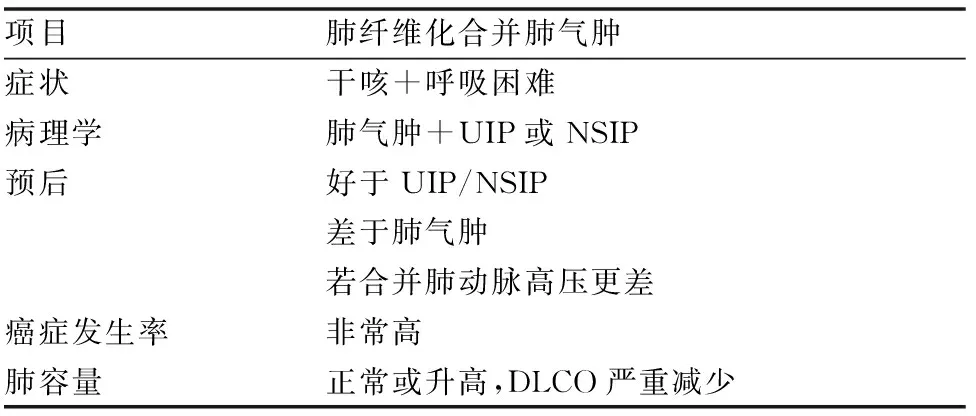
表2 肺纤维化合并肺气肿基本特征
CPFE 的治疗与单纯肺气肿或肺纤维化的患者相似,患者均应停止吸烟,并长期评估氧疗的适应症,并筛选出适合肺移植的患者。通常情况下,肺功能指标下降率在单纯纤维化患者和CPFE患者之间有所不同。CPFE患者FVC和DLCO随着时间推移下降较慢,相对于IPF患者不同,IPF患者的FEV1/FVC比率增加,CPFE患者的这一比率可能会降低。虽然在IPF中FVC、DLCO每年下降率是疾病生存率的独立预测因子,但CPFE患者的生存率与FEV1的下降密切相关[43, 46]。预后主要是由肺动脉高压决定,肺动脉高压发生在47%的CPFE患者中,并且与第1年内60%患者死亡率相关[44-46]。
总 结
吸烟会导致呼吸道和肺实质的多种变化。除了对肺泡壁的破坏导致肺气肿之外,还能通过多种内源性和外源性生长因子共同促进纤维化环境的产生,以此揭露吸烟是诱导这些疾病产生的关键因子。RB-ILD、DIP、PLCH、AEP、SRIF、CPFE组成的疾病谱统称为吸烟相关性间质性肺疾病(表 3),我们需从临床表现、组织病理、影像特征等多方面入手进一步研究这些疾病的关联。因此,无论 ILD 属于哪种类RB-ILD:呼吸性细支气管炎-间质性肺炎;DIP:脱屑性间质性肺炎;PLCH:肺朗格汉斯细胞组织细胞增多症;AEP:急性嗜酸性粒细胞性肺炎;SRIF:吸烟相关性特发性肺间质纤维化;CPFE:肺纤维化合并肺气肿综合征型,当我们对其他潜在疗法的作用不断深知时,戒烟仍然是这类疾病患者的主要且首要的建议。

表3 吸烟相关性间质肺疾病比较
