头颈部鳞癌免疫相关lncRNA的生物信息学分析
2022-05-20吴嘉雯唐加山
吴嘉雯,唐加山
(南京邮电大学 理学院,南京 210023)
0 前 言
头颈部癌症位居世界恶性肿瘤第六位,2020年全球头颈部癌症新增病例约93万人,死亡病例约47万人[1],其中约90%的病例被归类为头颈部鳞癌(HNSCC)。[2]尽管当前在治疗方式上取得了重大进展,但HNSCC患者的5年生存率并没有得到明显的提高。[3]近年来随着肿瘤免疫微环境领域的发展,有研究表明HNSCC患者的预后与肿瘤组织免疫微环境密切相关。[4,5]这提示我们可以识别免疫相关的预后指标,以改善HNSCC患者的预后并指导他们的治疗。
长链非编码RNA(ln cRNA)被定义为大于等于200个核苷酸的RNA,目前还没有证据表明它们可以被翻译成肽。随着研究的深入,ln cRNA已经被证明在肿瘤的发生和发展中起着不可或缺的作用。[6,7]研究表明,其中与免疫相关的ln cRNA在癌症的预后中具有独特的价值,免疫相关ln cRNA作为新兴的癌症生物标志物已被用于多种癌症的诊断和生存预测。例如,Shen等人[8]在乳腺癌中识别了11种免疫相关ln cRNA并构建预后标志物,并且证明了这个标志物和免疫细胞亚型的浸润有关;Li等人[9]在肺腺癌中识别了7个与免疫相关的ln cRNA并构建稳健的预测模型,以提高肺腺癌的预后预测。同样在HNSCC中,Wu等人[10]整合了与临床数据相关的ln cRNA、microRNA和mRNA,并建立了一种新的免疫相关RNA特征来预测HNSCC患者的存活率;Yin等人[11]基于13个免疫相关ln cRNA对来建立HNSCC的预后模型,但在这项研究中仅基于训练集数据进行了模型的构建和验证。因此,本文旨在提出一种新的基于免疫相关ln cRNA的生存预后模型来预测HNSCC的预后,并在训练集和测试集上对构建的模型进行验证。
本文获取了HNSCC患者的ln cRNA表达数据和相应的临床信息,将样本随机分为训练集和测试集。基于训练集,筛选得到HNSCC中具有预后价值的免疫相关ln cRNA,建立预后风险评分模型;并在训练集和测试集上对模型的性能进行了验证。结果表明,基于7个免疫相关ln cRNA构建的预后模型可作为一个独立的预后因素用于HNSCC的生存预后预测,并且这7个ln cRNA可能成为HNSCC的潜在治疗靶点。
1 材料与方法
1.1 数据来源及处理
HNSCC患者的转录组数据来自癌症基因图谱数据库(The Cancer Genome Atlas,TCGA)(https://portal.gdc.cancer.gov/)中的HNSCC项目组,并从中提取了lncRNA表达数据;HNSCC患者的临床病理信息来自UCSC Xena(https://xenabrowser.net/)。将HNSCC患者的lncRNA表达数据和临床数据进行整理和合并,使得整理后的样本均有对应的表达数据和临床数据,并删除生存信息缺失和生存时间小于30天的样本。
1.2 免疫相关lncRNA的提取
R软件中的edgeR是基于负二项分布的统计方法,根据样本的ln cRNA表达量,选择FDR<0.05和|log2FC|≥1作为阈值筛选出在肿瘤样本和非肿瘤样本之间有明显差异表达的ln cRNA。另外从免疫学数据库(Immunology Database and Analysis Portal,ImmPort)(https://www.immport.org/home)网站获得免疫相关基因列表,采用Pearson相关系数计算并确定与免疫基因相关的lncRNA。
1.3 预后模型的构建
首先,对免疫相关lncRNA进行单因素Cox回归分析,筛选出与总生存期(OS)显著相关的ln cRNA(P<0.01)。然后,建立Lasso回归模型,并进行10折交叉验证确定最优模型来进一步筛选出关键的ln cRNA。最后利用多因素Cox回归模型进行ln cRNA的筛选,并在建模过程中根据简单可行的Akaike信息准则(AIC)采用逐步回归法选择最佳模型。根据多因素Cox回归分析中加权线性组合的回归系数β和ln cRNA的表达量构建风险评分预后模型并利用模型来确定每个患者的风险评分。风险评分模型一般具有如下的形式[8,9]
rs=βln cRNA1×exprln cRNA1+βln cRNA2×exprln cRNA2+…+βln cRNAn×exprln cRNAn
根据风险评分的中位数将患者划分为高险组和低风险组。
1.4 预后模型的验证
采用Kaplan-Meier方法绘制生存曲线,并通过Log-rank检验来比较高低风险组的生存是否具有差异;通过单因素和多因素Cox回归分析进行独立预后分析;并绘制时间依赖性ROC曲线和计算ROC曲线下的面积(AUC值)以验证模型的预测效果。
2 实验结果
2.1 数据处理
从UCSC Xena网站下载了612名HNSCC患者的临床信息,并将临床信息和基因表达数据进行匹配,得到539个匹配样本;其中包括495个肿瘤样本和44个正常组织样本;去除生存信息缺失和生存时间小于30天的样本,最终得到486个样本。
根据基因注释文件,从HNSCC的全基因转录组数据中得到15 878个ln cRNA的表达数据,通过差异表达分析(FDR<0.05,|log2FC|≥1)得到1 729个差异表达的ln cRNA。根据ImmPort数据库下载的免疫相关基因信息,从转录组数据中提取了1 279个免疫相关基因的表达数据;采用Pearson相关分析筛选与免疫相关基因相关的lncRNA,结果确定了988个免疫相关ln cRNA(|R|>0.4,P<0.001)。
2.2 关键免疫相关ln cRNA的筛选和预后模型的构建
按照3:2的比例将样本随机分为训练集和测试集。在训练集中,对包含988个ln cRNA的表达数据进行了单因素Cox回归分析,以确定具有预后差异的免疫相关ln cRNA。从分析中发现,有20个免疫相关ln cRNA与HNSCC患者的OS相关(P<0.01)。再基于这20个ln cRNA建立Lasso回归模型,通过10折交叉验证确定最优模型,选择使模型达到最小损失时的惩罚参数λ(λ=0.019),得到加入模型的14个ln cRNA。接着对这14个ln cRNA,根据AIC值进一步建立多因素Cox回归模型,该模型的C指数为0.7。最终,将筛选得到的7个免疫相关ln cRNA纳入风险评分模型,其相应系数见表1。风险评分模型如下:
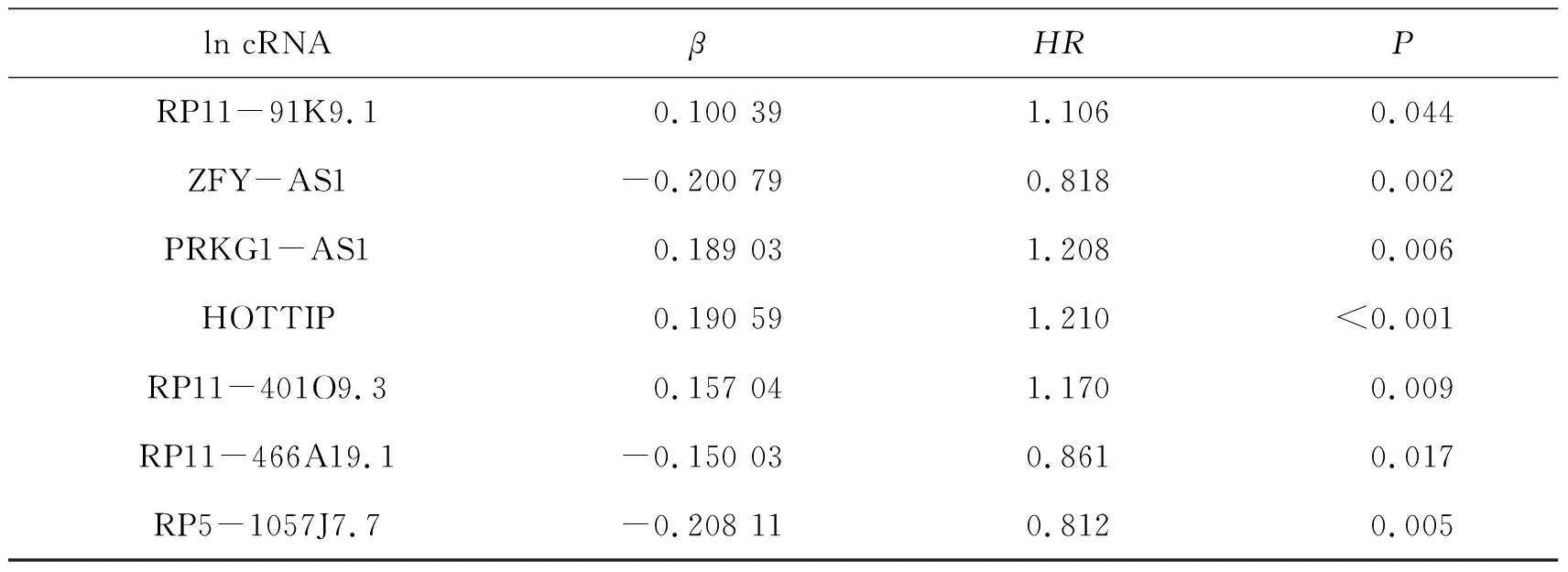
表1 多因素Cox回归分析筛选出纳入预后模型的免疫相关ln cRNA
风险评分=[RP11-91K9.1表达量*(0.100 39)]+[ZFY-AS1表达量*(-0.200 79)]+[PRKG1-AS1表达量*(0.189 03)]+[HOTTIP表达量*(0.190 59)]+[RP11-401O9.3表达量*(0.157 04)]+[RP11-466A19.1表达量*(-0.150 03)]+[RP5-1057J7.7表达量*(-0.208 11)]。
2.3 预后模型的验证
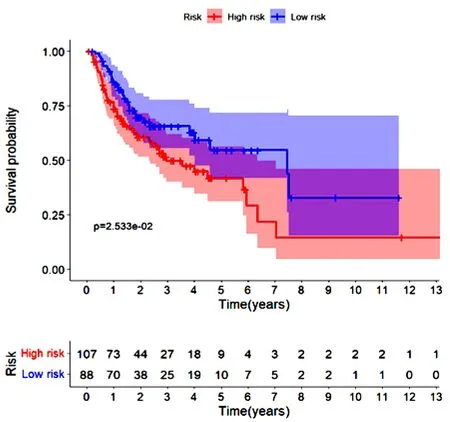
在训练集上,根据上述风险评分模型计算出每个病人的风险得分,并根据风险评分的中位数将训练集中风险评分大于中位数的患者归为高风险组,反之则归为低风险组(图1(a));所有病例的生存状态分布见图1(b)。热图(图1(c))显示了高风险组和低风险组之间7个ln cRNA的表达水平,这7个ln cRNA表达水平的变化趋势与风险评分计算公式中对应的系数一致。风险评分随着RP11-91K9.1、PRKG1-AS1、HOTTIP和RP11-401O9.3的表达水平的增加而增加,随着ZFY-AS1、RP11-466A19.1和RP5-1057J7.7的表达水平的增加而减少。Kaplan-Meier生存分析结果显示,低风险组患者3年总生存率比高风险组患者高约30%,5年总生存率高约20%(图1(d))。此外,绘制了时间依赖性ROC曲线来评估模型的预后价值,计算得到3年和5年总生存率AUC值分别为0.770和0.688(图1(e))。为了进一步评估生存预后模型的性能,在测试集中进行了相同的分析,并且得到了类似的结果,结果见图2。考虑到其他临床特征变量(如年龄、性别、分期和分级),进行了单因素和多因素Cox回归分析来验证风险评分可作为HNSCC的独立预后因素。单因素Cox回归表明,风险评分与较差的生存率相关;多因素Cox回归分析结果显示,基于7个关键ln cRNA的风险评分是HNSCC患者的独立预后因素(P<0.05)(图3)。综上可知,该模型具有一定的生存预后预测能力。

(a)风险评分的分布
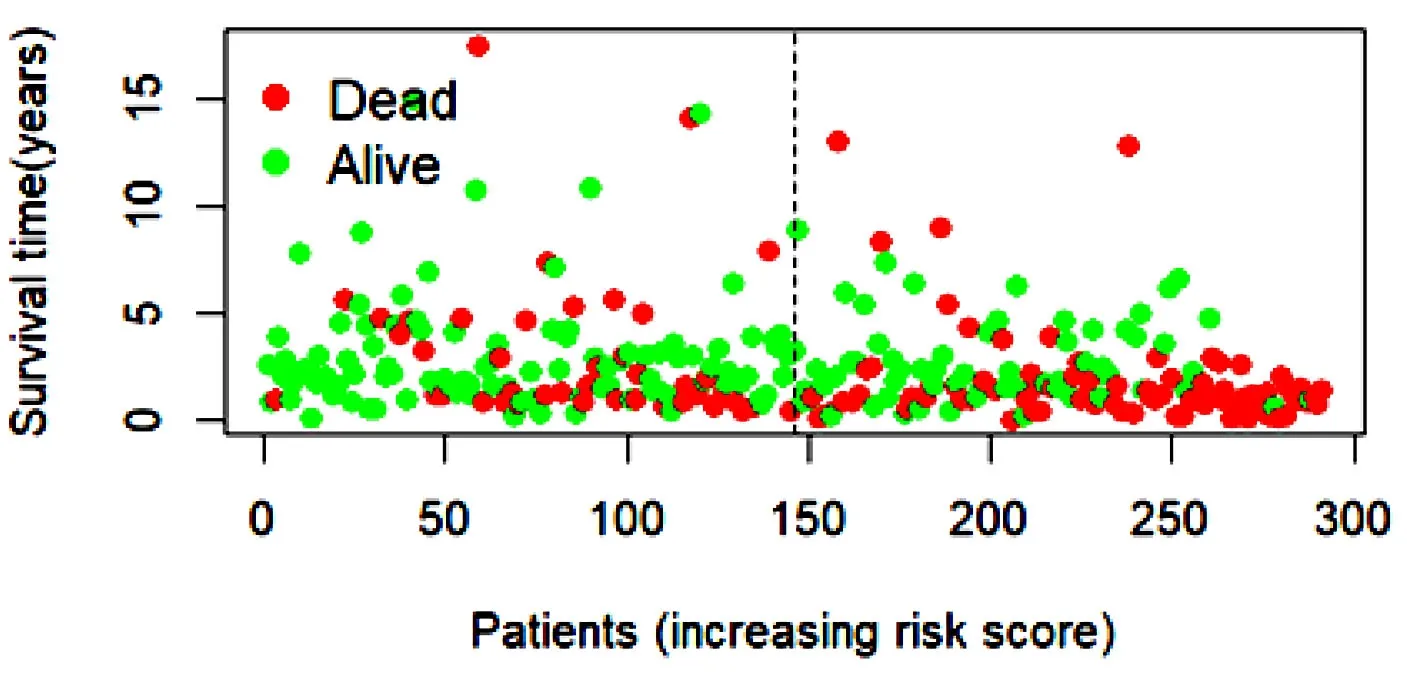
(b)生存状态和时间

(c)7个ln cRNA表达水平的热图

(d)高风险组和低风险组患者的Kaplan-Meier生存曲线 (e)风险评分的ROC曲线图1 训练集预后风险评分模型的构建
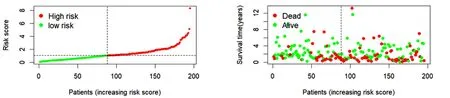
(a)风险评分的分布 (b)生存状态和时间
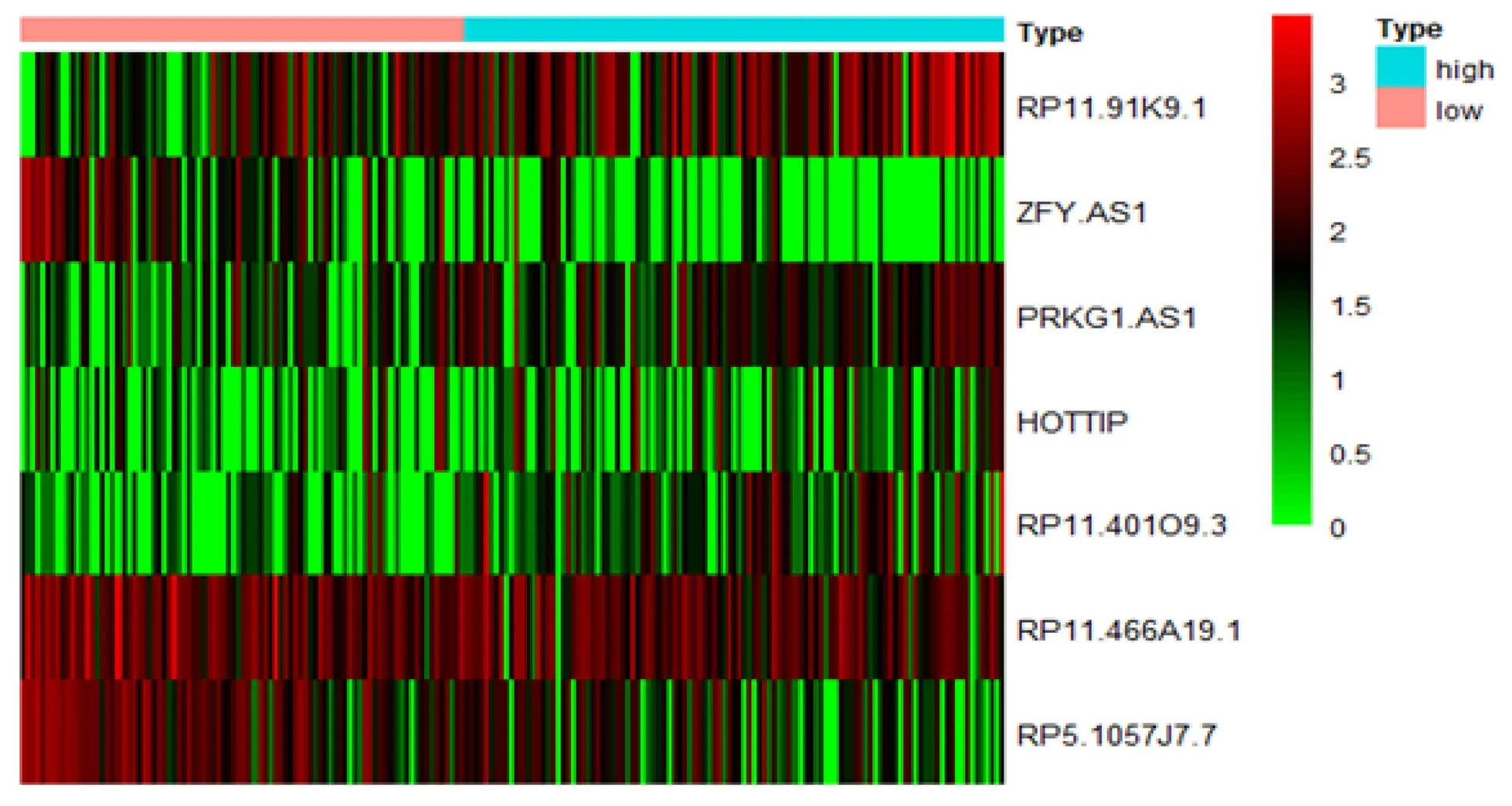
(c) 7个ln cRNA表达水平的热图
(d) 高风险组和低风险组患者的Kaplan-Meier生存曲线
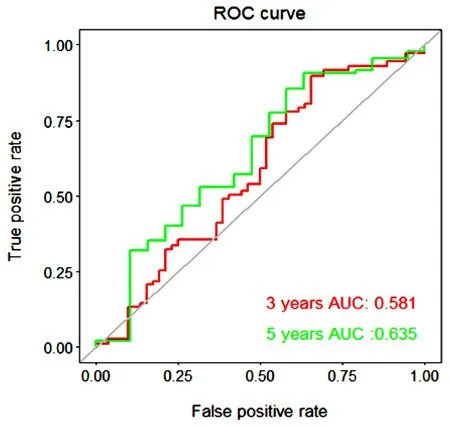
(e)风险评分的ROC曲线 图2 测试集预后风险评分模型的验证

(a)训练集的单因素Cox回归分析 (b)训练集的多因素Cox回归分析
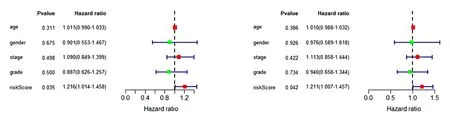
(c) 测试集的单因素Cox回归分析 (d) 测试集的多因素Cox回归分析

(e)整个数据集的单因素Cox回归分析 (f)整个数据集的多因素Cox回归分析图3 风险评分和临床特征的Cox回归分析
3 讨 论
近年来,ln cRNA成为肿瘤研究的热点,人们发现ln cRNA在肿瘤的发生和发展中起着重要作用,ln cRNA的异常表达被认为是影响肿瘤活性的关键因素;其中与免疫相关的ln cRNA被证明与恶性肿瘤的不良预后有关。[12,13]因此,我们的研究目的是通过对TCGA数据库中HNSCC患者数据的生物信息学分析,确定与HNSCC预后相关的免疫相关ln cRNA。通过对HNSCC患者的相关数据进行单因素Cox回归分析、Lasso回归分析和多因素Cox回归分析,我们确定了7个关键的免疫相关ln cRNA(RP11-91K9.1、PRKG1-AS1、HOTTIP、RP11-401O9.3、ZFY-AS1、RP11-466A19.1、RP5-1057J7.7),建立了用于HNSCC生存预后的风险评分模型,并根据风险评分的中位值将HNSCC患者划分为高风险组和低风险组。Kaplan-Meier曲线显示,基于模型划分的高风险组和低风险组的生存率存在差异且具有统计学意义(P<0.05)。同时在训练集和测试集中,模型5年生存率的ROC曲线的AUC值分别为0.688和0.635,表明该模型具有较好的预测能力。另外在筛选得到的7个ln cRNA中,ln cRNA HOTTIP被认为是几乎所有类型癌症中的致癌ln cRNA,也是人类恶性肿瘤中公认的生物标志物和治疗靶点[14];ln cRNA PRKG1-AS1被证明在口腔鳞癌的细胞生长、迁移和侵袭中起促进作用[15]。而其他ln cRNA目前没有相关的报道,还需要进行进一步的探索。
本文基于对TCGA数据库中HNSCC患者数据的分析,确定了7个与预后相关的免疫相关ln cRNA,并成功构建了一个用于HNSCC患者预后评估的风险评分模型。这个预后模型能在一定程度上反应HNSCC患者的预后情况,并且这7个免疫相关ln cRNA有望成为HNSCC潜在的免疫治疗靶标。
