RBP结合位点预测的深度学习方法进展
2022-05-18董正心潘小勇沈红斌
董正心, 潘小勇, 沈红斌
(上海交通大学 电子信息与电气工程学院,上海 200240)
RNA结合蛋白(RNA binding protein,简称RBP)作为细胞中重要的蛋白质之一,是RNA代谢的关键组成部分。它在基因调控过程中发挥着重要作用,包括转录后的剪接、加工、修饰、转运等RNA代谢过程,影响着RNA的结构并相互作用[1]。随着高通量测序技术的发展,RBP的更多功能逐渐被发现。大量实验结果表明,RNA结合蛋白在影响染色质水平上存在着广泛的功能和作用,各种不同的RBP可通过调控RNA来控制转录,增强RNA与蛋白质之间的相互作用,且RBP可作为转录因子或辅助因子来调控转录的[2]。例如,在环状RNA的形成过程中,RNA结合蛋白QKI起到了重要的调控作用,它通过结合pre-mRNA(不均一核RNA)上的特定结合位点来促进环状RNA的形成[3]。此外,遗传学数据和大量蛋白质组学数据表明,RBP与神经系统疾病、癌症等许多人类疾病有关,由RBP异常引起的RNA代谢缺陷可能是许多人类疾病的基础[4]。因此,通过研究RNA与RNA结合蛋白之间的相互作用的信息来揭示RBP的调控机制,对于探索RNA功能、治疗疾病等都有着重要意义。
目前,分析RNA与蛋白质相互作用或者定位RBP结合位点最常用的方法为高通量测序技术,如交联免疫共沉淀结合高通量测序(cross linking immunoprecipitation and high-throughput sequencing,简称CLIP-seq)[5]、RNA结合蛋白免疫沉淀结合高通量测序(RNA binding protein immunoprecipitation and high-throughput sequencing, 简称RIP-seq)[6]等。由于细胞中的RNA与RBP结合会形成核糖核蛋白(RNP)复合物,高通量测序技术首先利用特定蛋白的抗体将对应的RNA-蛋白质复合物沉淀下来,再分离复合物提取其中的RNA,并对其进行测序。高通量测序技术在疾病基因筛查以及人类基因组学研究等方面做出了突出贡献[7]。但对于规模庞大、类型复杂的基因组测序数据,这种通过人工筛选来确定RBP结合位点的方法,不仅测序实验时间长,价格也非常昂贵,其测序结果的准确性也依赖于实验环境及实验器材。因此,如何使用基于数据驱动的方法来研究RNA和蛋白质的相互作用成为一个有吸引力的研究方向。随着近年来大数据技术的发展,机器学习方法蓬勃发展,在生物信息领域得到了广泛应用,使用计算机对海量的测序数据进行模式挖掘的方法也快速发展。
1 RNA-蛋白质结合位点数据库
随着高通量测序技术和生物信息学的发展,关于RNA与蛋白质相互作用及RBP结合位点的大型数据库层出不穷。其中,大部分数据库不仅整合了大量来自如CLIP-seq等技术的实验数据,而且通过独立实验对RBP的准确性进行了验证和筛选,具有较高的可信度,为基于数据驱动的方法如机器学习方法提供了数据基础,用来构建可靠的基准数据集并用于机器学习模型的训练、测试、验证及评估。表1列举了近几年发表的可用于查询RBPs结合位点及其他RNA与蛋白质相互作用信息的部分数据库。
表1中,RBPDB[8]是一个RNA结合特异性数据库,它收集了所有的具有已知RNA结合位点域的RBPs结合实验数据,实验物种包括人、小鼠、蝇和蠕虫,数据涵盖272种RBPs、71个模体的位置权重矩阵和36套来自免疫沉淀实验的体内结合转录序列。

表1 RNA-蛋白质结合位点数据库
通过对来自37个独立研究的108个CLIP-Seq数据集进行解码,starBase[9]建立了RNA-RNA和蛋白质-RNA的相互作用网络,不仅可以进行泛癌分析,还可以执行部分RNA的存活和差异表达分析。CLIPdb[10]构建了一个RBP-RNA相互作用的数据库,包含各种高分辨率的RBPs在RNA上的结合位点及人工注释,可以在全基因组尺度上直观地显示出RBPs的结合位点。由于RBPs和miRNA的联合作用可认为是形成了一种转录后调控编码,DoRiNA[11]数据库主要用于分析转录后调控过程中RNA的相互作用。类似地,RBP-Var[12]数据库提供了RBP在转录后调控及相互作用的功能变体的注释,可以判断单核苷酸变异体(SNVs)是否可以影响RNA的二级结构,并识别可能会被破坏结合的RBPs,因此,可用于探索人类疾病背后的SNVs。ATtRACT[13]是RNA结合蛋白和相关模体的数据库,可以发现在一组序列中重复出现的模式,并与已存在的模体进行比较。特别地,RBPTD[14]数据库涵盖了与人类癌症相关的RBPs数据,通过整合28种癌症的基因表达谱、预后数据和DNA拷贝数变异(CNV)等数据来研究RBPs的变异原因和潜在功能。RBP2GO[15]提供了一个全面的RNA 结合蛋白数据库,包括从人类到细菌等13个物种的信息,105种RBPs,并且允许对具有特定分子功能的RNA结合蛋白进行反向搜索。另外,POSTAR[16]系列数据库基于高通量测序数据,主要探索了7个物种的转录后调控机制,提供了最大的带功能注释的RBPs结合位点的集合。
2 编码方法
对于机器学习模型来说,需要输入数值型的数据来保证模型内部的计算。因此,对于非数值型数据如由碱基组成的RNA序列,在输入模型前需要进行合理的编码使其转化为数值数据。这个过程称为特征编码,一般在输入模型前得到的是初级的稀疏特征,而机器学习模型可以从中提取有用的信息得到高级的抽象特征,从而完成特定的机器学习任务。以下介绍3种在RBP结合位点预测领域常用的几种特征编码方式。
2.1 独热(one-hot)编码
独热编码常用于离散型数据的编码,这种数据常包含多个类别,且各个类别的重要程度没有明显差别。经过独热编码后可得到由单个1和多个0组成的N维向量,其中N由类别数决定,1所在的位置由当前类别决定。编码成向量形式同时也意味着将数据映射到了欧式空间,各个类别初始的独热编码特征向量之间的距离相同,而经过模型学习后的高级特征之间的距离会发生变化,即体现了当前任务中各维特征的重要程度。对于用于RBP结合位点预测的RNA序列,包含A、C、G、U(或T)4种碱基,因此经过独热编码可以得到L×4的独热矩阵,其中L为序列长度。这也是最常用的RNA序列编码方法,其编码过程如图1所示。

图1 独热编码示例
2.2 K-mers
在生物信息学中,K-mer是指对于输入的序列数据进行切分产生的长度为K的序列片段。如对于RNA序列来说,即为包含K个碱基的RNA子序列。K-mers方法常用于基因组复杂度分析、基因组组装等,还可以通过生成K-mers谱结合一些概率模型来研究基因组的分布,如低阶马尔可夫模型[17]。基于K-mer的编码方式主要有2种。以k为3为例,共有64种3-mer片段,对于每条RNA序列生成64维编码向量。一种方法是对于输入的每条序列切分生成3-mer片段集合,统计集合中每种3-mer出现的次数即为该条序列的64维编码向量对应维度上的特征数值,也称基于K-mer频率的编码方法,其编码过程如图2(a)所示。另一种方法借鉴了自然语言处理领域的方法,将K-mers片段集合当作单词词袋,单个K-mer视为单词,RNA序列视为句子,考虑了K-mers之间即RNA前后序列之间的连续关系,可用词嵌入方法生成对应的嵌入编码进行进一步的学习。另外,基于分子生物学的特征也有一些编码方法的拓展。例如,在信使RNA分子中,相邻的3个核苷酸可在蛋白质翻译时对应一种氨基酸,可视为K为3时的一个特例。如CRIP[18]方法将所有的64种3-mer片段对应20种氨基酸和1个终止密码子进行映射,得到3-mer片段的21维独热编码的特征向量,相较于4维独热编码得到了更丰富的序列信息,其编码过程如图2(b)所示。

图2 K-mers编码方法
2.3 RNA二级结构
除了对RNA的序列信息进行编码之外,RNA的二级折叠结构也蕴含着大量信息,因此可以将RNA二级结构转化为拓扑图结构。常用的RNA二级结构预测方法有RNAFold[19]、MFold[20]、RnaPredict[21]等,如RNAFold预测具有最小自由能的RNA二级结构。如RPI-Net[22]方法在得到预测的RNA二级结构后,把碱基作为图上的结点,把序列主链及二级结构中的碱基配对信息作为图中连接的边,得到相应的拓扑图结构,其过程示意图如图3所示。
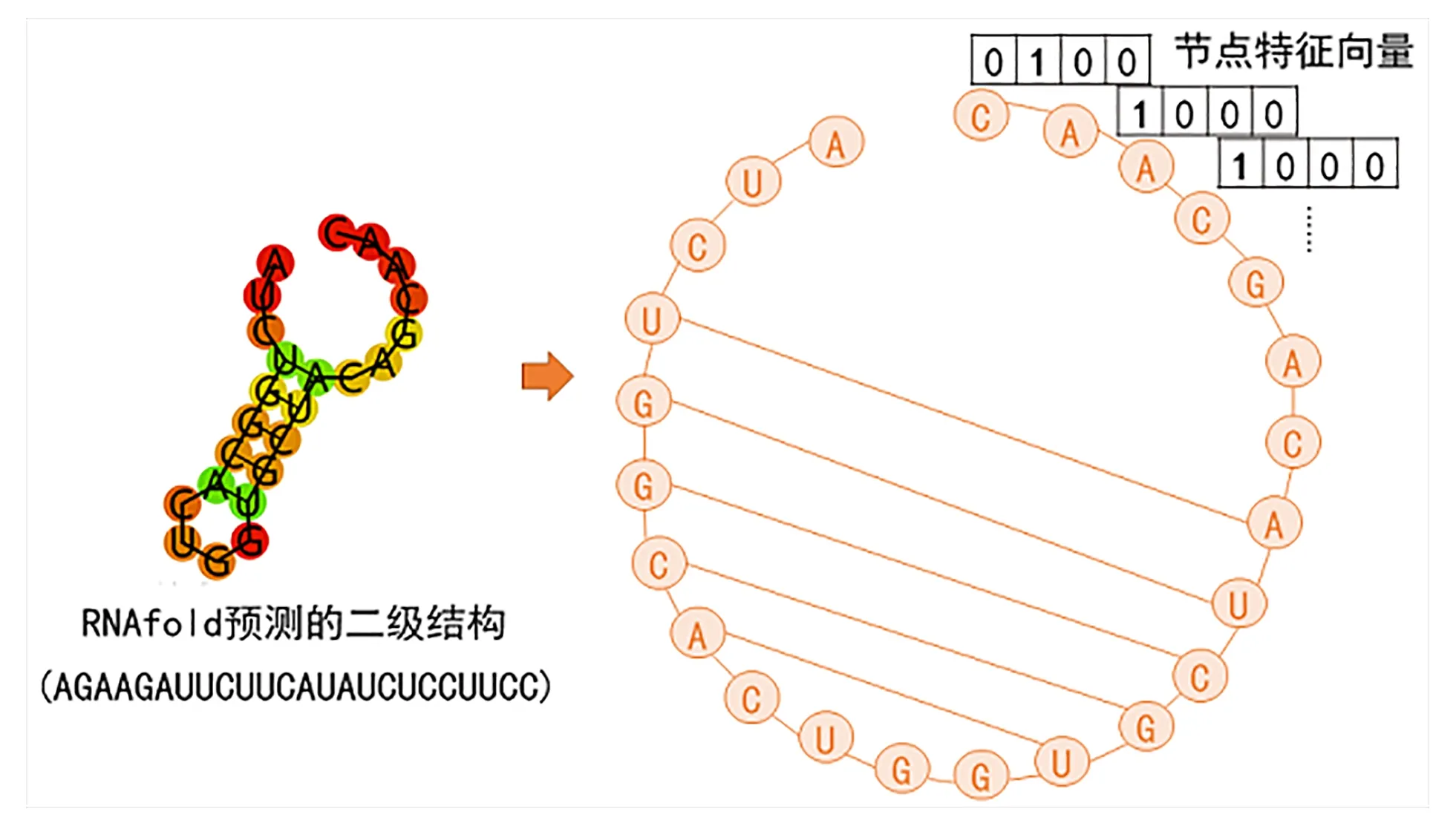
图3 RNA二级结构
3 机器学习
在过去的几十年里,机器学习在多个领域得到了广泛的应用并取得了优越的成果,如计算机视觉、自然语言处理、生物信息学等。机器学习方法可以从训练数据的已知样本中学习到隐含的模式,并用于对未知样本的预测。大部分机器学习方法为有数据标签的监督学习模型,如支持向量机、随机森林等,而非监督学习没有数据标签信息。这里的传统机器学习指浅层结构的模型,深度学习指具有深层结构、包含多层神经网络的模型。
3.1 传统机器学习
3.1.1 支持向量机
支持向量机(support vector machine,简称SVM)[23]是经典的机器学习技术之一,常用于解决各种分类与回归问题。SVM是一种广义线性分类器。除了线性分类外,SVM还可以通过核函数将非线性输入向量映射到线性高维特征空间,并在该特征空间中构造一个最大间隔超平面,该超平面以最大间隔将两类数据分开。在机器学习中,SVM常使用带正则项的铰链损失函数(hinge loss function),其学习目标为类间几何间隔最大化即损失函数最小化,因此求解SVM可以看作求解二次凸优化问题,常用的方法有梯度下降、坐标下降、内点法等。
由于SVM较强的泛化能力,其应用也越来越广泛。不仅可以进行光学字符识别,还可用于文本及图像分类,并且越来越广泛地应用于生物学中,如微阵列基因表达谱分类、蛋白质分类等[24]。
3.1.2 随机森林
随机森林(random forest,简称RF)[25]是决策树的集合,其中每棵树都依赖于独立采样的样本训练数据,其泛化误差取决于每棵树的性能和彼此之间的多样性,且随着决策树数量的增加收敛到一个极限值。RF在训练过程中进行随机特征选择,通过投票法或平均法聚合多棵决策树的预测结果来得到最终的预测结果,这种随机抽样和集成策略使得它具有较高的预测精度和泛化能力。
由于RF在处理高维特征空间和复杂数据结构上具有独特的优势,其在生物信息学领域的应用也越来越广泛[26]。如利用肿瘤标志物表达训练RF来检测肿瘤样本簇[27],或利用RF来预测蛋白质对之间的相互作用[28]。
3.2 深度学习
3.2.1 多层感知机(MLP)
多层感知机(multilayer perceptron,简称MLP)[29]是一种前馈人工神经网络,可以通过不同的激活函数将一组输入向量映射到一组输出向量。MLP一般由3层或者更多层网络构成,其基本结构包括输入层、隐含层、输出层,每层都由多个节点组成,每个节点即一个带有激活函数的神经元。在训练过程中,各神经元的权重参数可以通过监督学习的反向传播算法更新,以减小实际值与预测值之间的误差。由于激活函数的可选择性,MLP可用于解决多种复杂问题。
3.2.2 深度置信网络(DBN)
深度置信网络(deep belief networks,简称DBN)[30]通常由多层隐含层构成,其层内单元没有联系,具有逐层学习的特点。DBN可以看作多个子网络的组合,如受限玻尔兹曼机(RBM)或自动编码器,其中每个子网络的隐藏层充当下一个子网络的可见层。DBN具有2个特征:1)自上而下学习的生成权,决定某层的变量如何根据上一层的变量来改变;2)自下而上传递的生成权,由底层的数据向量推断每层潜在变量的值。DBN可用于处理高度结构化的数据(如图像),且经过训练后可以用来初始化隐含层进行数据特征降维。由于其处理复杂数据的优势,DBN被应用在许多领域,如预测单声道隐马尔可夫模型状态的概率分布[31]。
3.2.3 循环神经网络(RNN)
循环神经网络(recurrent neural networks,简称RNN)[32]是一种带有循环结构的神经网络,它通过隐藏状态来存储过去的信息,再与当前输入一起决定当前输出及隐藏状态的更新,常用于处理序列数据,如自然语言等。由于RNN在训练过程中出现参数爆炸、梯度消失等问题,且无法解决长期依赖问题,难以学习到较长时间之前的信息,因此引入了多个变种,如长短期记忆单元(long short-term memory,简称LSTM)、门控循环单元(gated recurrent unit,简称GRU)等。LSTM[33]由输入门、遗忘门、输出门及记忆单元等构成,这种结构使LSTM在训练过程中可以决定输入的信息是否应该保留或遗忘,解决了长期依赖问题,适用于长时滞后的任务,可用来处理较长的序列数据。GRU[34]是LSTM的一个改进版本,它对门控机制进行了改进,其单元内部只包括重置门和更新门。
3.2.4 卷积神经网络(CNN)
卷积神经网络(convolutional neural networks,简称CNN)属于前馈神经网络,一般由卷积层、池化层、全连接层组成。卷积操作为使用一定大小的卷积核在输入特征上进行滑动,并将感受野内的数值与卷积核进行对应元素的乘法运算,然后将乘法结果进行加和得到卷积后特征图中的对应元素值。其中卷积核对应参数由训练确定,在训练过程中使用反向传播算法来优化卷积核参数。池化操作是一种降采样方法,常采用最大池化、平均池化等方法对上一层输出的特征图进行降维操作。经过多个卷积层和池化层,可以提取到高级抽象特征,再经过一层或多层全连接层完成相应的分类或回归任务。
CNN目前已在多个领域广泛应用,如图像分类、目标检测、语义分割、自然语言处理等,近年来也常用于药物发现,如AtomNet[35]将卷积概念应用于生物活性和化学相互作用的建模,使用三维卷积来预测小分子生物活性。由于CNN强大的特征学习能力,可以挖掘出数据中蕴含的空间结构,CNN也可用于模体的挖掘,对于研究和理解RNA与蛋白质相互作用有重要意义。
3.2.5 图神经网络(GNN)
图神经网络(graph neural networks,简称GNN)[36]是一种处理图数据的网络,可用于多种图类型数据,如无向图、有向图、无环图、循环图等,将图及其节点映射到欧几里得空间进行运算。类似的领域有图嵌入,它旨在学习节点或图的低维、稠密的特征表示,同时保留图的拓扑结构和节点内容,如Deepwalk[37]使用随机游走的方法获取局部信息来学习节点的潜在嵌入特征表示。使用深度学习的方法来进行图嵌入学习时也可归于图神经网络领域。GNN领域中图卷积网络(graph convolutional network,简称GCN)占有重要地位,包括谱方法和空间方法。通常GCN的输入包括节点特征矩阵和图结构描述性矩阵,如邻接矩阵,GCN可以进行端到端的学习,其示意图如图4所示。

图4 图卷积神经网络(GCN)示意图
近年来,随着图神经网络的发展,提出了各种变体网络,如GraphSAGE[38]通过对邻居节点采样、聚合的操作实现以节点为中心的小批量训练来代替全图训练;GAT[39]将注意力机制引入图神经网络,通过注意力机制聚合邻居节点并自适应地分配权重,提高了表达能力。除了应用在社交网络、知识图谱、推荐系统等领域外,图神经网络在生物信息学上的应用也越来越广泛,包括小分子结构、基因/蛋白质相互作用网络等,如GCNG[40]利用图神经网络将高通量空间表达数据中的空间信息编码为图,并结合表达数据来推断基因相互作用。
4 RBP结合位点预测
目前,大部分预测RBP结合位点的方法将预测问题转化为分类问题,来判断蛋白质是否能与RNA某些区域绑定。对于RNA序列,能与蛋白质绑定的区域称为结合位点,其他区域称为非结合位点。因此,可以训练一种蛋白质特异性的二值分类器来预测RNA上的RBP结合位点,该分类模型需要RNA的表征作为输入,训练数据通常使用高通量测序技术获得。对于特定蛋白质,需要收集足够多的训练数据来预测RBP在RNA上的绑定位点。由于不同的RBP具有不同的绑定模式,对于每种RBP需要单独训练一个模型。
4.1 基于传统机器学习预测RBP结合位点的方法
传统的机器学习方法需要通过特征工程清洗数据、提取特征,以及使用更浅层的学习模型,如SVM、RF等。表2总结了近年来使用传统机器学习来预测RBP结合位点的方法。

表2 基于传统机器学习预测RBP结合位点的方法
RNAContext[41]方法提出了基于结构上下文字母表来标注RNA序列的单个碱基,集成了序列和结构信息来推断RBPs的绑定倾向,可以准确预测短序列上RBP的绑定强度,学习到结合位点的3 D形状的更精确描述。之后,RCK[42]在RNAContext的基础上进行了改进,使用了一种新的基于K-mer的模型,同样用到了结构信息,将RNAplfold预测的结构上下文的概率向量与序列一起作为输入。基于K-mer的方法能更好地引入上下文信息,但同时也意味着模型参数增多,提高了模型过拟合的风险。
GraphProt[43]方法则首次利用完整的二级结构信息,采用了一种高效的图-核方法,将预测得到的结构编码成图,其中包含序列和完整的二级结构信息,之后再使用图核的方法从图中提取特征,最后通过训练SVM模型对RBP结合位点进行分类。在RBP-24数据集的评估上,GraphProt在24组中有20组的表现都优于RNAContext。实验结果表明[43],加入结构信息相比不加结构信息使得模型在平均相对误差上有大幅度下降,因此,模型的准确性能得到了提升,但是模型训练速度下降。
与前几种方法不同的是,Oli[44]方法仅使用了序列特征作为模型输入,提取RNA序列的K-mer频率作为输入特征,用SVM作为分类器对RNA-蛋白质的相互作用进行分类。另外,该方法同时提出了基于模体评分的OliMo[44]方法以及基于二级结构的OliMoSS[44]方法。经过实验证明[44],仅基于序列的Oli方法通过四核苷酸特征表示提取到了足够的结合特性,这时结构信息可能是不必要的。
iONMF[45]使用了一种正交矩阵分解方法来整合多个数据源,旨在发现类别特异性的RNA结合模式,其中,蛋白质与RNA相互作用的关键预测因子是RNA序列和结构模体的位置、RBP的共结合及基因区域类型。该方法验证了融合多个数据源的模型比在单个数据源上取得了更高的准确性,而且这种正交正则化非负矩阵因子分解的方法给数据集成技术提供了新的方案。
RNAcommender[46]考虑到蛋白质结构域的组成和RNA预测的二级结构,利用了相互作用信息,针对RBPs的RNA靶标训练了一个推荐系统,为未知的RBPs预测RNA靶标。
综上所述,可以看出基于传统机器学习的方法注重于数据特征的处理、选择和集成,而加入额外信息的作用与模型本身有关。例如,在Oli方法中,二级结构特征未起到提升预测性能的效果,但在iONMF方法使用的多源数据特征中,又验证了信息量最大的数据源是RNA结构。因此,当前模型是否能有效利用到结构信息也是需要考虑的因素。对于基于K-mer频率的方法,在Oli方法中证实了4-mer核苷酸的方法能有效提取到结合特征,虽然RCK方法中指出这种方法会导致参数的增加,需要交叉验证来避免过拟合,但是在模型训练速度上并未下降,因此,基于K-mer的方法是一种值得考虑的有效的特征提取方法。
4.2 基于深度学习预测RBP结合位点的方法
随着实验验证的RBP结合位点数据的迅速积累,可以使用深度学习挖掘出越来越多隐含在数据里的绑定模式。深度学习被广泛地应用在计算机生物学,如用来预测RBP结合位点的深度学习模型针对每种蛋白质训练一个蛋白质特异性模型。表3对于近年来在RBP结合位点预测领域上提出的基于深度学习的方法进行了总结。
4.2.1 基于RNA序列预测RBP结合位点
经过多种方法证明,仅使用RNA序列作为模型输入也可以提取到足够信息,取得较好的预测效果,下面介绍几种基于RNA序列预测RBP结合位点的深度学习方法。
DeepBind[47]是第一个使用卷积神经网络来预测RBP结合位点的方法,并且可以利用卷积层学习到的参数来挖掘序列绑定模体,获得的准确性优于基于传统机器学习的方法。DeeperBind[48]则在卷积神经网络的基础上添加了一个LSTM层来学习序列内的长期依赖信息,结合深度学习特征表征的能力,使用高通量技术产生的数据训练模型学习序列的结合特性,进一步提升了预测性能。DanQ[49]使用了类似的CNN和双向LSTM的网络架构来学习调控机制,预估突变的影响,其预测染色质水平的能力意味着它可以更好地预测遗传变异引起的表观遗传变化。由于RNA序列的转录机制、序列长度等特点,CNN通过训练卷积核在局部感受野内提取高级抽象特征,而LSTM则凭借其学习长依赖信息的能力很好地处理了较长的RNA序列,使得CNN与LSTM组合的网络取得了优越的性能。
类似于CNN与LSTM的组合网络结构,MSCGRU[50]是一种结合多尺度卷积层和双向门控递归单元(GRU)层的预测模型。多尺度卷积层能够捕获不同长度的模体特征及RNA-蛋白质之间局部的结合模式,双向GRU层能捕获子序列之间的依赖关系,从而预测RBP的绑定基序。
一般来说,由局部和全局序列决定RBP是否能与某序列片段进行绑定。因此,iDeepE[51]结合了一个局部多通道CNN和一个全局CNN来预测RBP结合位点,其中局部CNN处理多个重叠的固定长度子序列,而全局CNN处理整条序列,且局部CNN在保持和全局CNN相似性能的情况下处理速度更快。由iDeepE的结果可知,更深的模型不意味着更好的预测性能,因为更深的模型往往意味着需要更多的训练样本来保证模型的泛化性能。经实验对比发现,只有2层CNN的模型比有20层CNN的ResNet表现得更好,特别是对于那些已知RNA靶点较少的蛋白质。在RNA长序列上iDeepE比DeepBind表现得更好,但在短的RNA序列片段上两者表现相似。
随着注意力机制在计算机视觉领域取得了可观的进展,它在其他领域上的应用也越来越广泛。iDeepA[52]引入基于注意力机制的卷积神经网络来自动搜索重要位置如绑定基序,并用来预测RBP结合位点。iDeepA中集成了CNN和2层注意力层,并提取了来自CNN和注意力层的3个输出特征图。对于具有少量已知RNA靶点的蛋白质,引入注意力机制提升了预测能力。但是对于具有大量已知RNA靶点的蛋白质,引入注意力机制不能提升预测能力。一个可能的原因是注意力机制可以快速定位到重要的基序,因此不需要更多的训练样本来学习高级特征。
另外,K-mer方法也可以编码RNA序列,但是K-mer频率不能对不同的K-mer之间的距离建模。考虑到核酸的多态性,一些K-mer在语义上是相关的。因此,一些方法首先将K-mer当作单词,将序列当作句子,使用词嵌入方法学习分布式特征,然后使用学习到的特征来分析K-mer之间的相似性。如iDeepV[53]引入了word2vec词嵌入方法,首先,从全基因组序列中学习K-mers的低维、稠密的分布式向量,然后将这些学习到的向量进一步输入CNN以区分结合位点与非结合位点。对于某些只有少量训练样本的RBP,iDeepV的表现优于DeepBind。另外,学习到的分布式特征可以用于其他的下游分类任务。与传统的K-mers方法相比,分布式表示特征对于检测K-mers之间潜在的相互关系和相似性更有效。类似地,RBPSpot[54]方法也使用了K-mer方法来搜索具有统计学意义的基序,结合其上下文信息来评估序列的绑定潜力。除了分布式特征外,一些传统的语言模型也可以用于特征提取。如kDeepBind[55]中使用k-Gram统计语言模型提取了序列的k-Gram频次,与CNN提取的特征拼接后输入全连接层完成分类任务。
综上所述,与基于传统机器学习的方法相比,得益于神经网络强大的特征提取能力及处理大型数据集的强大的计算能力,基于深度学习的方法在仅有序列信息的情况下可以取得更好的预测性能。如DeepBind方法仅使用CNN模型,但在RBP-24数据集上的评估AUC达到了0.92,高于RNAContext和GraphPort,在RBP-31数据集上的评估AUC达到了0.85,与iONMF持平,但也高于Oli和GraphProt。
4.2.2 引入结构信息预测RBP结合位点
RBP也通过识别结构上下文与RNA靶点结合,因此,结构信息也被用来预测RBP结合位点和结合偏好。多种方法证明,添加多源特征能提升性能,特别是一些互补的特征。考虑到RBP结合位点的异构表示及除序列外其他影响RNA-蛋白质相互作用的因素,iDeep[56]方法通过集成包括区域类型、共绑定、结构概率、模体得分和RNA序列共5种多源数据特征来学习其之间的共享特征。其中使用CNN捕捉序列中的调控模体,使用DBN学习其他4种数据输入的隐藏状态的高级特征,2个模型独立预训练后,再结合在一起进行最后的联合训练。iDeep在性能上超过了其他先进的方法,并且可用于推断绑定序列模体。从iDeep的结果可看出,区域类型和共绑定信息对于预测RNA上的RBP结合位点起到了较大作用,而且使用多模态的方法也能从各模态中学习到更有效的特征。
类似地,考虑到调控序列不仅取决于核酸序列,还取决于其与基因组标志物,如转录起始位点、外显子边界或聚腺苷酸化位点的相对距离,CONCISE[57]引入样条变换,构建了一种基于样条函数的神经网络模型,针对各基因组标志物之间的相对距离建模。由CONCISE的结果可知,其性能超过了其他基于距离的机器学习模型。但局限性在于,输入的标量特征的尺度需要提前确定,以保证样条节点均匀地分布在整个特征值范围内。
在进行RBP结合位点预测时,常伴随着挖掘模体的任务。如iDeepS[58]使用2个CNN和一个双向LSTM来同时学习结合序列模体和结构模体,还可以学习到序列和结构间的长期依赖信息。iDeepS仅使用了序列及根据序列预测的结构,由于iDeep使用了其他多源的特征,例如基因组背景信息,在一些RBP上iDeepS表现得比iDeep差,但与其他基于序列和结构的方法如GraphProt相比,iDeepS表现更好。Pysster[59]能通过CNN同时检测序列和结构中的模体,其中,序列和结构通过组合序列和结构的字母表得到的扩展字母表来进行编码,并对输入序列分类。
随着预测RNA二级折叠结构方法的进步和准确性的提升,一些二级结构预测结果可作为可靠数据加入输入特征。如DLPRB[60]根据RNAplfold预测得到RNA结构上下文的概率向量,对结构信息进行特征编码,再使用CNN和RNN从高通量体外数据中联合分析RNA序列和结构,其网络结构新颖性在于,RNN在RNA结合位点预测中的应用及CNN中数百个可变长度卷积核的组合。cDeepBind[61]通过轻量级CNN用于转录组范围推理和适用于小批量数据的LSTM,将计算预测的二级结构特征作为模型输入,并证实了其在提高预测性能方面的有效性。
除了学习RNA序列的分布式表示外,DeepRKE[62]利用RNA一级序列和二级结构的分布式表示来推断RNA结合蛋白结合位点,使用了非监督的浅层神经网络,使用嵌入词算法提取RNA序列和二级结构的特征,即分布式表示的K-mers序列,然后将学习到的序列分布式特征输入CNN和双向LSTM来预测RBP结合位点。Deepnet-rbp[63]则首次考虑了RNA三级结构信息,将序列、二级结构、三级结构信息编码为统一的特征表示,描述了RBP在所有3个维度上的结构特异性,然后输入多DBN模型来预测RBP结合位点和模体。其中,使用了RNAshapes工具预测可能的二级结构,利用一种复制式softmax模型对原始序列和二级结构进行编码,并通过JAR3D工具预测序列的三级结构模体。实验结果表明,整合额外的RNA三级结构特征可以提高模型在预测RBP结合位点方面的性能。
除了将RNA序列表示成独热编码,RNA二级结构也可以表征成图。GraphProt2[64]首先通过RNAfold预测序列的二级折叠结构,然后以此为基础将预测的二级结构编码为图。使用碱基作为图中的节点,将其one-hot编码及保守性分数、区域类型等特征作为节点特征,使用碱基配对信息建立图中的边,得到了完整的无向拓扑图网络,然后利用图神经网络模型预测RBP结合位点。与GraphProt不同的是,GraphProt2对整条RNA序列计算了位置预测得分,预测性能优于iDeepS。
与仅使用序列的方法相比,将预测的结构引入模型训练可以在一定程度上提高预测性能,但也大大增加了计算量,对于长RNA序列来说非常耗时。结构信息包含二级折叠结构、结构概率、区域类型等多种信息,如何结合模型结构有效利用多种结构信息,如GraphProt2中结合图神经网络模型提取RNA二级折叠结构的特征,是进一步提升的关键点。
4.2.3 多标签、多任务、多模态
与上述针对每种RBP训练一个模型的RBP特异性模型不同,iDeepM[65]提出了一种多标签深度学习方法。iDeepM将预测RBP的问题建模成多标签的分类问题,通过一个CNN和一个LSTM来预测绑定的蛋白质,其中CNN用来提取高级基序特征,而LSTM用来学习RBP之间的长期依赖关系。iDeepM的一个优势在于不需要为模型训练构建负样本集,而是利用了RBP之间的依赖关系。多模态深度学习可以学习到多源数据的共享高级特征,而对于RBP绑定的RNA,每个模态都有其自身的表征。对于RBP结合位点预测,这些共享的高级特征具有强大的辨别能力。类似地,DeepRiPe[66]构建了多任务、多模态的DNN模型,使用模块化结构从DNA/RNA序列和转录本区域类型中学习信息特征,然后将这些模块的特征合并输入多任务模块,同时预测多个RBP的结合位点。因此,DeepRiPe模型既能够使用任务之间的共享信息,又能专注于每个RBP的独特特征。
这种多标签、多任务、多模态的结构相较于单任务学习关注到了更多的关联信息,考虑到了不同的RBP之间的相似性及相互作用,在多个任务之间共享学习到的信息,为单个RBP的预测提供了补充。
4.2.4 非编码RNA
近年来,除了信使RNA(mRNA)以外,对其他非编码类RNA分子的探索也成为热门的研究方向,如环状RNA(circRNAs)、长链非编码RNA(lncRNA)等。非编码RNA通常指不翻译蛋白质的RNA,但这并不意味着这些RNA不包含信息或不发挥功能[67]。实际上,这些非编码RNA也通过某种模式来控制基因表达的水平,参与了与其他核酸和蛋白质相互作用的复杂网络,对细胞生物学具有广泛的影响,并在疾病中扮演重要角色。在癌症研究中,非编码RNA已被确定为多种癌症的致癌驱动和肿瘤抑制因子,其相互作用的失调导致了肿瘤的发生,并揭示了重要的新靶点[68]。
环状RNA(circRNAs)是一种具有共价闭合结构、高稳定性的RNA,参与基因调控,由线性RNA的5’端和3’端经共价结合形成[69]。在CRIP[18]方法中,使用了基于密码子编码的方法,通过结合卷积神经网络和长短时记忆网络来研究环状RNA与RBP的相互作用,从而预测RBP在circRNA上的结合位点。iCircRBP-DHN[70]方法利用深层次网络识别circRNA与RBP的结合位点,首先提出了一种新的编码方法CircRNA2Vec,旨在通过一种无监督文档嵌入方法从circRNA序列中捕获长距离依赖关系,然后和k元组核苷酸频率模式结合起来表示不同程度的核苷酸依赖性。该网络结构可看作一个具有自注意机制的双向门控递归单元(BiGRUs)的深层多尺度残差网络,它能同时提取局部和全局上下文信息。该模型不仅表现出在识别环状RNA-RBP相互作用位点方面的潜力,而且在线性RNA数据集RBP-31上也表现出了优于CRIP、iDeepS的性能。iDeepC[71]是一种RBP特异性方法。它采用了一个由轻量级注意力模块和度量模块组成的孪生神经网络。其中,孪生神经网络通过成对度量学习有效地提高了网络捕获环状RNA之间互信息的能力,在一定程度上解决了部分RBPs的已知绑定circRNA数量有限的小样本问题。类似地,长链非编码RNA(lncRNA)一般指长度超过200 bp的一类RNA[72]。HOCNNLB[73]方法通过一种基于高阶核苷酸编码的卷积神经网络模型来预测RBP在lncRNA上的结合位点,在长链非编码RNA上表现出了优秀的预测性能。
4.3 性能比较
为了比较各方法的性能,对比了不同模型在RBP-24[43]和RBP-31[45]两个数据集上的公开实验结果。
RBP-24数据集来自GraphProt (http://www.bioinf.uni-freiburg.de/Software/GraphProt/),由21个蛋白质的24个实验组成,其中23组来自doRiNA[74],另外一组是PTB的HITS-CLIP结合位点数据[75]。其训练和测试序列具有可变长度,且不同RBP的训练样本数量不同。
RBP-31数据集来自iONMF (https://github.com/mstrazar/ionmf),是一个序列具有固定长度的基准数据集。其中,每条序列的长度为101 nt,每个RBP有24 000个训练样本、6 000个验证样本和10 000个测试样本。它由24个蛋白质的31个实验组成,在每个实验中,首先确定cDNA计数最高的核苷酸位置作为正样本备选池,然后在距离小于15个核苷酸的位置中,只考虑具有最高cDNA计数的位置当作正样本以避免冗余,负样本取自在任何实验中都未检测到相互作用的基因中的序列区域。
此处只比较实验中报告的平均接收者操作特征曲线(receiver operating characteristic curve,简称ROC)下的面积(AUC)。ROC曲线由预测结果和真实标签共同决定,其纵坐标为真阳率(TPR),横坐标为假阳率(FPR),计算方法为:
(1)
(2)
其中:TP指预测为正类的正样本;FP指预测为正类的负样本;TN指预测为负类的负样本;FN指预测为负类的正样本。因此,TPR可以理解为模型正确预测的能力,而FPR为模型错误预测的程度,当TPR越高时,曲线下面积越大,即AUC越高,模型性能越好。
由各方法公开的数据可知,基于深度学习的方法通常比传统的基于机器学习的方法表现更好。深度学习广泛用于预测RBP结合位点的蛋白质特异性方法,基于深度学习的方法比基于浅层学习的方法具有更好的性能。可能的原因是,蛋白质的样本数非常大,这对训练深度学习模型非常有利。
对于在RBP-24上的实验数据,DeepBind方法凭借卷积神经网络强大的特征提取能力,表现出了优于RNAContext与GraphProt的预测能力。与DeepBind具有相当性能的iDeepA、iDeepV方法的平均AUC同样达到了0.92,证实了注意力机制与词嵌入方法应用在RBP结合位点预测领域上的有效性。同时,融合局部CNN与全局CNN的iDeepE方法的AUC达到了0.93,也展现了CNN在关注不同范围内重要信息的能力。带预训练的孪生网络iDeepC的平均AUC达到了0.94,表现出了最高的准确性,同时适用于小样本的蛋白质预测任务,在一定程度上缓解了由于部分RBP的已知RNA靶点不足带来的问题。
对于在RBP-31上的实验数据,DeepBind方法同样表现出了优于Oli、GraphProt及iONMF方法的预测能力,基于深度学习的方法显示出了更多的优势。特别是,与使用多个特征源的基于矩阵分解的iONMF方法相比,仅使用序列的深度学习模型产生更好或相当的性能,并且iONMF的平均AUC达到了0.85,而同样具有多源特征的基于多模态深度学习的CONCISE方法和基于多尺度卷积网络的MSCGRU方法的平均AUC达到了0.92,展现了深度学习网络在融合多源数据、多尺度特征上的强大能力。
4.4 模体(motif)挖掘
模体是普遍存在于核酸或蛋白质等生物大分子中的保守序列,可看作序列集合中的一种公共序列模式,或具有特定功能的序列片段。在蛋白质中表现为一种具有特定功能的超二级结构,包括线性短模体、结构模体等。模体挖掘对定位生物序列中有意义的序列片断起着重要作用。RNA结合蛋白的序列特异性表现出深度的进化保守性,可以从RBP的RNA结合域序列推断其绑定偏好,因此,模体挖掘对于分析人类转录后调控机制,探索RBP与RNA靶标之间的序列特异性关联具有重要意义。深度学习方法已经被广泛应用在RBP绑定模体挖掘上,特别是卷积神经网络,检测模体一般是基于卷积神经网络预测RBP结合位点方法的副产物,可理解为通过CNN学习到RBP结合序列的高级特征。表4、表5分别对近几年挖掘序列模体和序列-结构模体的方法进行了总结。由iDeep可知,可以把学习到的卷积核参数转化为位置权重矩阵,以匹配输入序列来挖掘RBP的绑定模体。另外,iDeepC方法基于可解释性方法来挖掘绑定模体,通过使用集成梯度来计算每个碱基对蛋白和RNA绑定的重要性贡献,得到序列上每个碱基的重要性分数,进而找到那些连续分数较高的序列片段,作为潜在的RBP绑定模体。

表4 序列模体挖掘方法

表5 序列-结构模体挖掘方法
5 讨论
由以上可以看出,深度学习技术已经在RBP结合位点预测领域得到了广泛应用,显著地提高了RBP结合位点与非结合位点的分类性能,且通过挖掘模体提高了模型方法的可解释性。但是,深度学习技术的应用还具有一些限制,其在生物信息领域上的应用有待进一步改进与提升,据此提出了以下可行的改进方向。
5.1 数据集质量的提升
随着高通量测序技术的发展,大量序列数据得到积累,使得深度学习技术的应用成为可能,但也决定了模型能达到的预测精度的上限。目前,由于测序技术上的局限性及实验的不确定性,数据仍存在假阳性和假阴性的问题。而且,大多数模型在处理序列时需要进行切割,将其中的包含结合位点的片段分离出来用作正样本。这个过程也会因为选取的分割方法不合适而破坏结构完整性、序列连续性,因此引入噪声或丢失信息。同样地,在构造负样本集时也会受到影响,并且负样本数远大于正样本数。另外,RBP数据集在不同的RBP之间也存在不平衡的问题,而部分RBP的数据集由于样本数较少,导致模型训练不充分而达不到更高的预测精度。因此,更高质量的数据集可能会更大地提升预测性能。
5.2 探索更多的深度学习模型
由各方法在RBP-24与RBP-31数据集上的公开实验数据可以看出,基于多通道CNN的iDeepE方法在RBP-24上的平均AUC达到了0.93,而在RBP-31上基于多尺度CNN的MSCGRU方法的平均AUC达到了0.92,都表现出了最好的预测能力。由此可见,这种融合多维度特征的深度学习方法能有效提取RNA序列上隐含的有用信息,从多个尺度学习到重要特征。同时,在MSCGRU方法中BiGRU网络也起到了学习长期依赖信息的重要作用,能够在全局的维度上学习不同尺度特征之间的相关性,这也使得网络更适用于RNA序列上的学习任务。因此,如何针对RNA序列及RBP结合位点的特点量身定制合适的深度学习网络,或者按照其特殊的结合模式提出独特的学习方法,是在RBP结合位点预测领域上进一步突破的关键。
近年来,除了介绍的几种深度学习模型外,更多改进的深度学习模型也层出不穷。迁移学习、强化学习、对比学习、多任务学习等也逐渐被应用到生物信息领域,如MTTFsite[81]是一个多任务学习框架,通过利用跨细胞系的数据来解决数据缺乏的问题,其中包含一个共享的CNN来学习所有细胞系中转录因子的共同特征以及一个私有的CNN来学习每个细胞系中转录因子的私有特征,特别是在那些标记数据不足的细胞类型上提升了预测性能,在一定程度上缓解了数据不均衡的问题。另外,深度学习模型相当于一个“黑匣子”,对于其在RBP结合位点预测上应用的可解释性,模体挖掘作为探索的第一步已经取得了较好的效果。未来,对RNA-蛋白质结合机制上更多的可解释性研究,需要进一步探索。
6 结束语
本文主要回顾了近年来深度学习在RBP结合位点预测领域上的预测方法进展。首先,总结了常用的RNA-蛋白质结合位点数据库。然后对应用于传统机器学习模型和深度学习模型的编码技术进行了介绍,主要是RNA序列的编码方法。接下来介绍了传统机器学习和深度学习技术的发展。进一步对近年来应用传统机器学习或深度学习技术来预测RBP结合位点、挖掘模体的方法进行了详细介绍,并对其在RBP结合位点数据集上的性能进行了比较。最后,讨论了目前深度学习方法应用上的局限性及其可能的改进方向。
