肠道结节性淋巴组织增生症36例临床病理特点分析
2022-05-10郭芷含计晓彬代晓艳史海磊武杰何冰
郭芷含 计晓彬 代晓艳 史海磊 武杰 何冰
(青岛大学附属医院病理科,山东 青岛 266003)
肠道结节性淋巴组织增生症(Nodular lymp-hoid hyperplasia,NLH)是一种以肠道结节性息肉样隆起为特征的良性病变,是以胃肠道黏膜分化良好的淋巴细胞增生、分裂活跃并形成弥漫性的淋巴滤泡为特点的一种疾病。本病比较罕见,国内文献报道大多为个案或小样本病例报道。肠道NLH通常伴发于普通变异型免疫缺陷病、获得性免疫缺陷症、肠道病毒感染等情况,患者通常因反复发作的、顽固的呼吸道、消化道及其他部位感染就诊,以儿童多见。同时,肠道NLH为淋巴瘤的高危影响因素,不论患者是否存在免疫缺陷,均容易发展成为肠道及肠外器官淋巴瘤。肠道NLH的诊断主要依靠内镜及病理学检查,病变单发或多发,以小肠、直肠等部位多见,形态学上黏膜层及黏膜下层内可见增生的淋巴组织,生发中心增生活跃。由于肠道NLH与部分肠道原发淋巴瘤形态学有相同或相似之处,需要通过完善的免疫组织化学检查及分子基因检测来进行鉴别。本研究收集36例肠道NLH患者的资料,结合文献复习对其临床病理特征、免疫表型、分子检测、鉴别诊断及患者预后情况进行总结,旨在提高病理医生对该疾病的认识,探讨该疾病的发生及与其他伴发疾病可能存在的关联性。
1 材料与方法
回顾性分析青岛大学附属医院2015年11月—2020年9月经内镜活检病理检查诊断为肠道NLH的36例患者的临床资料,包括患者年龄、性别、临床症状、是否有幽门螺旋杆菌(HP)感染、内镜表现、其他相关疾病等。通过电话形式进行随访。
同时收集整理36例肠道NLH患者活检组织标本的形态学表现及免疫组织化学检测结果(包括CD20、CD3、bcl-2、bcl-6、CD10、CD5、CD79α、CD21、CD23、cyclinD1、Ki67)。对所有患者标本进行免疫球蛋白(Ig)(IgH、IgK、IgL)基因重排检测:从石蜡包埋组织样本中提取基因组DNA,使用欧洲BIOMED-2方案和ABI 3500DX型基因分析仪对标本基因组DNA进行检测分析,检测位点包括IgH的FR1-JH、FR2-JH、FR3-JH、DH-JH、DH7-JH片段,IgK的Vκ-Jκ以及Vκ-Kde+intron-KKde片段,IgL的Vλ-Jλ片段。
2 结 果
2.1 一般资料
36例肠道NLH患者中男30例,女6例;年龄11~66岁(中位年龄30岁);5例为未成年人。28例因腹痛就诊,其中4例伴有腹泻;5例因便血就诊,其中1例出现明显的体质量下降;另外1例于体检时无意发现,1例因胆囊癌术后嗳气行内镜检查时发现,还有1例11岁男性患者因高级别B细胞淋巴瘤化疗后行PET/CT检查时发现。其中5例伴有高血压,2例伴有恶性肿瘤(1例为胆囊癌,1例为高级别B细胞淋巴瘤),1例伴有嗜酸性粒细胞增多症,1例伴有结核杆菌感染,1例16岁男性患者伴有黑斑息肉(Peutz-Jeghers)综合征及轻度贫血。36例患者中15例进行了HP检测,其中5例伴有HP感染。36例患者随访时间为3~59个月,1例因恶性肿瘤死亡,其余35例均生存良好。
2.2 内镜检查结果
36例患者的发病部位分别为回肠末端26例,直肠5例,横结肠2例,乙状结肠1例,十二指肠1例,另外1例62岁男性患者累及回肠、升结肠及乙状结肠等多个部位。内镜下均可见黏膜表面息肉样隆起,单发或多发,直径0.2~0.5 cm,质软,无蒂或有短蒂(图1A)。11例在其他部位发现息肉,经过组织学检查证实,8例为管状腺瘤,2例为增生性息肉,1例Peutz-Jeghers综合征患者同时伴发回盲部的Peutz-Jeghers错构瘤性息肉以及直肠乙状结肠交界的管状腺瘤。
2.3 病理组织形态学特征
36例患者的活检组织标本中,镜下可见肠黏膜固有层淋巴组织明显增生,淋巴滤泡结构存在并形成息肉样小结节(图1B),其形态类似正常淋巴结但无包膜和淋巴窦。增生的淋巴滤泡大小不一,大部分淋巴滤泡的生发中心明显扩大,增生活跃,部分发生相互融合(图1C),围绕滤泡生发中心的套区境界清楚,高倍镜下见套区的淋巴样细胞排列紧密,大小、形态相似,可见有丝分裂,但无明显异型性和病理性核分裂象。
2.4 免疫组织化学特征
增生的淋巴滤泡CD20及CD79α呈强阳性表达(图1D),周边副皮质区及滤泡间区可见散在CD3以及CD5阳性的T淋巴细胞;生发中心细胞表达CD10及bcl-6,不表达bcl-2(图1E),Ki67在生发中心的增殖指数约90%;CD21及CD23染色显示淋巴滤泡内滤泡树突网完整(图1F),淋巴滤泡不表达cyclinD1。
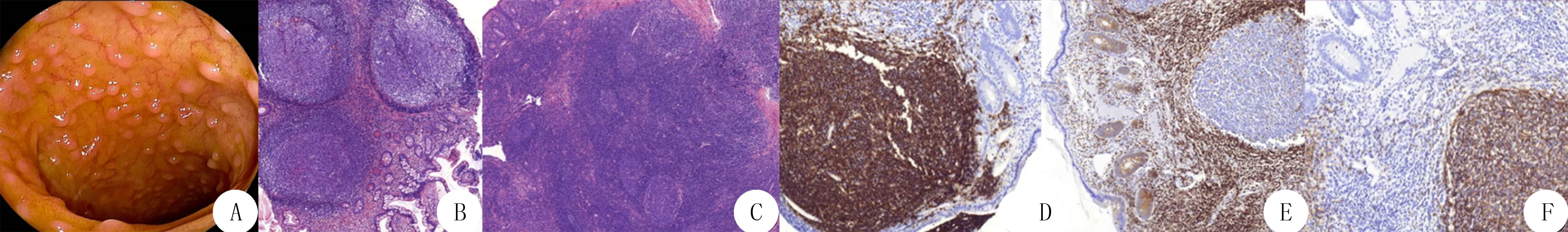
A:内镜检查显示黏膜表面多个无蒂息肉样隆起;B:肠黏膜固有层淋巴组织增生,存在淋巴滤泡结构,HE染色,100倍;C:生发中心明显扩大,部分相互融合,套区境界清楚,HE染色,100倍;D:增生的淋巴滤泡强阳性表达CD20,EnVision法,100倍;E:增生的淋巴滤泡生发中心bcl-2不表达,EnVision法,100倍;F:CD21染色显示淋巴滤泡内滤泡树突网完整,EnVision法,100倍
2.5 Ig基因检测结果
36例患者中19例进行了Ig(IgH、IgK、IgL)基因重排检测,结果均为阴性,未发现单克隆性B淋巴细胞。
3 讨 论
肠道NLH又称肠道淋巴样息肉病,是发生在肠道的一种少见的良性病变,可发生在人群各个年龄阶段,临床上多见于儿童及青少年,成年人较少见[1]。LIN等[2]研究显示,肠道NLH有明显的男性性别倾向,但RUBIO-TAPIA等[3]研究显示该病以女性多见,在本研究所收集的36例患者中,成年人31例,患者年龄与已报道文献存在一定差异,可能与就诊医院年龄构成有关。该病大多数患者以腹泻、腹痛为首发症状,部分患者也可能于体检中无意发现[4-5]。
肠道NLH大体上可表现为圆形、大小不等的小结节,色浅质软,数目不等,可以为单发也可多达数十枚。大多息肉表现为无蒂或宽基底,少数有粗短的蒂部。息肉表面黏膜多光滑,少数表面可有浅表糜烂形成。肠道NLH最常发生于回肠末端,也可发生于肠道其他部位[3,6-7]。本组研究中,26例发生于回肠,其他位于直肠、横结肠、乙状结肠及十二指肠,另外1例累及回肠、升结肠及乙状结肠等多个部位,与此前相关报道结果基本一致。
肠道NLH具有较为典型的组织学特征:息肉表面被覆相对正常的薄层黏膜,黏膜与黏膜下层之间形成单个或数个界限较清楚的次级淋巴滤泡,生发中心明显扩大,围绕生发中心的淋巴套区及周围边缘区相对境界清楚,高倍镜下见构成淋巴细胞大小、形态相似,无异型性。在滤泡之间可见淋巴细胞、浆细胞、嗜酸性粒细胞等浸润。肠道NLH病灶形态非常类似于小肠中的派尔斑及结直肠中的淋巴小结,而且较大的派尔斑在内镜下也可见到呈息肉样的外观。在形态上,派尔斑和淋巴小结表现为黏膜相关淋巴组织的散在分布,一般跨越黏膜肌层分布,由4个部分组成:淋巴滤泡、上皮下穹顶、滤泡间区及滤泡相关上皮,其内的淋巴滤泡以B细胞为主,另含有滤泡树突细胞和巨噬细胞,可见生发中心形成,滤泡间区富含T细胞。在功能上,派尔斑及淋巴小结属于胃肠道黏膜免疫系统的组成部分,是机体固有免疫防护的器官化淋巴组织,而肠道NLH可能是在免疫缺陷的情况下,为了弥补肠道淋巴组织的功能不足,由B淋巴细胞发育中的成熟缺陷导致浆细胞前体积累的结果[3,8]。
另外,部分发生在肠道的淋巴瘤与肠道NLH在内镜、影像学及形态学表现方面难以区分,还需依靠免疫表型及分子检测进一步进行鉴别。肠道NLH与以下淋巴瘤鉴别的要点为:①肠道结外黏膜相关淋巴组织(Mucosa-associated lymphoid tissue,MALT)边缘区B细胞淋巴瘤:MALT淋巴瘤通常伴有“淋巴上皮病变”,可见肿瘤性淋巴细胞侵犯黏膜上皮;肠道NLH中的滤泡结构比MALT淋巴瘤中的更为明显;同时大部分MALT淋巴瘤病例存在Ig重排。②套细胞淋巴瘤:发生于肠道的套细胞淋巴瘤可具有类似于肠道NLH的淋巴息肉样形态,但套细胞淋巴瘤表达cyclinD1及SOX11,CCND1基因转位是其遗传学特征,可与肠道NLH鉴别。③十二指肠型滤泡性淋巴瘤:可发生在十二指肠和回肠末端,内镜下表现为多发丘状或颗粒状隆起,镜下黏膜层见多个淋巴滤泡增生,相当于低级别滤泡性淋巴瘤的形态,十二指肠型滤泡性淋巴瘤存在IgH/bcl-2基因重排,免疫组织化学染色显示肿瘤细胞表达bcl-2[9],而肠道NLH不表达bcl-2,两者也可通过Ig重排鉴别。本研究36例患者中19例进行了Ig基因重排检测,结果均为阴性,提示Ig基因重排检测可作为肠道NLH和肠道淋巴瘤鉴别诊断的方法之一。
通常认为免疫因素在肠道NLH的发生过程中发挥重要作用,肠道NLH可发生于免疫缺陷、低丙种球蛋白血症和反复感染的患者[10-12],特别是与贾第鞭毛虫的感染相关[13-14],这突出了肠道淋巴组织的内源性功能的作用。现研究显示HP感染与肠道NLH有关[8],患者根除HP后病变明显消退[3]。本研究36例患者中,5例胃镜检查伴有HP感染,肠道NLH与HP感染间的关系尚需进一步研究;3例外周血检查分别显示调节性淋巴细胞亚群异常、嗜酸性粒细胞增多以及结核杆菌感染,这提示肠道NLH可能与免疫缺陷相关。有研究结果显示,肠道NLH患者还可能合并恶性肿瘤以及胃肠道间质瘤等[2,15]。本研究36例中有2例合并恶性肿瘤,1例合并Peutz-Jeghers综合征, 11例患者存在组织学类型不同的息肉,其中8例为管状腺瘤。结直肠腺瘤是结直肠腺癌主要的癌前病变[16],有研究提示一些非外生型结直肠腺瘤可能起源于淋巴组织相关的黏膜,并可以进一步发展为早期结直肠癌[17],但肠道NLH与结直肠腺癌之间的确切关系尚不清楚。本研究中8例肠道NLH合并管状腺瘤的病例随访期间尚未发现癌变。
通常认为肠道NLH为自限性疾病,可自行消退或经局部切除可治愈[18]。研究表明肠道NLH与肠外淋巴瘤的发生发展及转归有关[15]。JONSSON等[19]曾报道1例同时患有胃大B细胞淋巴瘤及肠道NLH的患者,在经过CHOP方案化疗后,增生的淋巴组织随着淋巴瘤的缓解而完全消失,在肿瘤复发时再次出现,但对于正常淋巴组织何时变为增生性或何时变为病理性增生,没有明确或有效的判断标准[19]。本研究中,1例11岁患儿因肠外(左扁桃体)高级别B细胞淋巴瘤行化疗期间,PET/CT检查显示肠壁增厚,内镜活检病理检查诊断为回肠末端NLH,并且外周血检查提示调节性淋巴细胞亚群异常。这种肠外淋巴瘤和肠道NLH之间的相关性目前并不清楚,推测可能是由于肠道内未知的抗原刺激,使得淋巴滤泡出现增生,也可能是副肿瘤综合征的一种表现。
综上所述,肠道NLH是一种极为罕见的肠道良性病变,诊断及鉴别诊断需要依靠内镜检查、病理组织形态学表现、免疫组织化学表达及基因检测结果,其发病机制尚不明确,可能与免疫缺陷、贾第鞭毛虫感染和HP感染等有关,并可能与一些恶性肿瘤的发生和转归相关。肠道NLH本身通常不需要特别干预,但其相关或可能伴随的其他肿瘤需要引起注意。
利益冲突声明:所有作者声明不存在利益冲突。
ConflictsofInterest: All authors disclose no relevant conflicts of interest.
作者贡献:郭芷含、何冰参与了研究设计、论文的写作和修改;计晓彬、代晓艳、史海磊、武杰参与了研究的实施。所有作者均阅读并同意发表该论文。
Contributions: The study was designed, drafted and revised byGUOZhihanandHEBing.JIXiaobin,DAIXiaoyan,SHIHailei, andWUJieparticipated in the implementation of the study. All the authors have read the last version of the paper and consented submission.
