PERK 通路对大鼠脑出血继发性脑损伤及神经细胞凋亡的作用
2022-05-05徐晨阳苑兵舰
徐晨阳苑兵舰
河南大学淮河医院 神经外科,河南 开封475000
脑出血(Intracerebral hemorrhage,ICH)带来较高的致死率和致残率,在全世界范围内均有较大的危害[1]。一般认为ICH 后血脑屏障(BBB)破坏引起的脑水肿和神经元凋亡是脑出血后继发性脑损伤的两大关键病理因素,产生原因包括血肿周围脑组织缺血和水肿加重,以及相关的毒性作用[2]。继发性脑损伤(Secondary brain injury,SBI)可引起细胞代谢紊乱,并激活应激反应,包括内质网应激(ER)和展开蛋白反应(UPR),这些应激反应要么重建细胞内稳态,要么激活细胞死亡程序[3]。
内质网应激是导致细胞凋亡的主要机制之一。错误折叠或展开蛋白的积累引起内质网功能障碍,从而引起内质网应激,随后它被诱导成信号传导级联(即UPR),细胞试图恢复体内平衡以防止死亡[4]。内质网激酶(protein kinase R,PKR)样内质网激酶(PERK)、激活转录因子6(activing factor 6,ATF6)和需要激酶1的肌醇(IRE1)促进了UPR 的形成。PERK 是一个中枢内质网应激传感器,它通过蛋白质转换和转录因子ATF4的诱导,迫使适应性程序恢复体内平衡,UPR 清除错误折叠的蛋白质可以促进神经元的存活[5]。因此,轻度内质网应激通过促进自噬发挥神经保护作用,但如果内质网应激持续或过度,则可能导致细胞死亡。后一种情况被证明对大鼠缺血或再灌注脑损伤的病理生理学有贡献[6]。此外,在神经退行性疾病中,抑制ER 应激可以抑制神经细胞凋亡[7]。CCAAT 增强结合蛋白同源蛋白(CHOP)是PERK 通路的下游效应物,作为促凋亡因子发挥作用。既往研究[8]表明,内质网应激激活caspase-12诱导细胞凋亡,但内质电应激和细胞凋亡是否与脑出血诱导的继发性脑损伤有关,其机制尚不清楚。本研究主要探讨PERK 信号在对脑出血继发性脑损伤及神经细胞凋亡的作用。
1 材料与方法
1.1 实验动物和实验材料
本研究选用普通的成年雄性SD 大鼠,体质量0.252~0.3 kg,购于中国科学院动物中心。将大鼠关在温度和湿度可控的动物房间,光照与黑暗各12 h。实验用抗体,均购自美国CST 公司。
1.2 体内与体外构建大鼠脑出血(ICH)模型
大鼠术前8 h禁食,不禁水。参照Tang Z P[9]的报道,于脑立体定位仪上采用缓慢注血法:前囟右侧旁开3 mm,进针深度5 mm,缓慢注射自体股动脉血50μL 至基底节区制成大鼠ICH 模型,注血时间约30 min。对照组不做注血操作。在体外,原代海马神经元用氧血红蛋白(Oxy Hb)处理。
1.3 实验分组
实验1:将48只大鼠随机分为6组,每组8只。取其中一组作为对照组ICHsham,其余5组按时间进程6 h、12 h、24 h、48 h、72 h 编 为ICH6h、ICH12h、ICH24h、ICH48h、ICH72h。大鼠在ICH后对应的时间点进行安乐死处理,分离脑组织并取其进行后续分析。
实验2:将48只大鼠随机分为6组,每组8只,取其原代海马神经元,在体外使用氧血红蛋白(Oxy Hb)处理。取其中一组作为对照组control,其余5组按时间进程6 h、12 h、24 h、48 h、72 h编为Oxy Hb6h、Oxy Hb12h、Oxy Hb24h、Oxy Hb48h、Oxy Hb72h。
实验3:将36只大鼠随机分为4组,每组9只,按处理方式编为sham、ICH、GSK2606414、salubrinal。
1.4 实验方法
采用免疫印迹(Western blot)、免疫荧光和TUNEL染色等方法,观察上述各组大鼠PERK 通路相关蛋白的表达及神经元细胞的凋亡情况。
1)免疫印迹。收集不同组的神经元细胞,根据细胞量加入适量RIPA 细胞裂解液,提取各组细胞的蛋白。配制体积分数为10%的SDS-PAGE,每孔加入30μg蛋白样品,经SDS-PAGE 电泳,转膜,用质量分数为5%的脱脂奶粉封闭2 h,TBST 稀释一抗(1∶1 000 稀释),4 ℃过夜;加入羊抗兔二抗(1∶2 000稀释),室温孵育2 h;ECL 发光。重复3次。主要抗体anti-CHOP 按体积比1∶2 000 稀释,anti-p-eIF2α、anti-eIF2αantibody、anti-XBP1、anti-caspase-12、anti-ATF-4、anti-ATF6、anti-β-Tubulin按体积比1∶1 000稀释。
2)免疫荧光检测。大脑组织在质量分数为4%的多聚甲醛中固定,嵌入在石蜡,切成4μm 部分,这是脱蜡前免疫荧光染色。然后,大脑部分染主要抗体,在4℃存放12 h。接下来,将大脑部分用PBS漂洗3次,沾染适当的二次抗体。免疫荧光分析采用正常兔IgG 作为阴性对照,切片用荧光法观察。主要抗体Neu N、antibody-neuronal细胞标记、antip-eIF2α、anti-ATF-4均按体积比1∶100稀释。
3)TUNEL 染色。采用末端脱氧核苷酸转移酶介导的d UTP Nick End Labeling(TUNEL)染色检测脑内细胞凋亡,具体步骤参照文献[10]。用荧光显微镜(尼康,日本)(每只大鼠3个切片)平行观察神经元并拍照。
1.5 数据统计分析
2 结果
2.1 Western blot法检测ICH 大鼠PERK 通路相关蛋白表达
在构建ICH 大鼠模型的过程中,检测处理后不同时间组中相关蛋白的变化。结果发现:在ICH 模型组中,随着时间的推移发现,p-eIF2α和ATF-4表达是逐渐增加的,且在48 h达到最顶峰;与ICHsham 组相比,其差异具有统计学意义(P<0.05),见图1A。此外,我们还确定了其他两个ER 应激通路相关基因的表达,其中ATF-6和XBP-1 的表达也是升高的,见图1B。
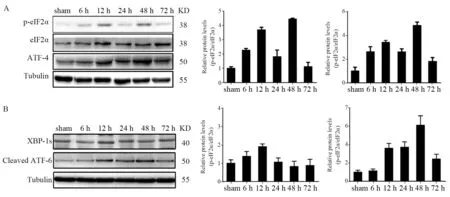
图1 构建lCH大鼠后,不同时间点大鼠中p-elF2α与ATF4蛋白质水平的变化
2.2 Western blot法检测Oxy Hb处理不同时间神经元中PERK 通路相关蛋白表达
在体外利用Oxy Hb处理神经元,模拟ICH 模型,检测处理后不同时间组中相关蛋白的变化。结果发现:在Oxy Hb处理组中,随着时间的推移,peIF2α和ATF-4表达是逐渐增加的,且在48 h达到最顶峰;与control组相比,其差异具有统计学意义(P<0.05),见图2A。在另外两个ER 应激通路中的ATF-6和XBP-1 表达也是升高的,见图2B。

图2 OxyHb处理神经元细胞不同时间后p-elF2α/elF2与ATF4蛋白的表达
2.3 免疫荧光检测ICH 处理48 h脑组织中eIF2α与ATF-6的表达
免疫荧光结果显示:与ICH sham 组相比,ICH后48 h,其脑部的p-eIF2α的蛋白质水平和ATF-4蛋白质水平都增加,这也证明了p-eIF2α和ATF-4主要在神经元中表达,见图3。因此,在后续的48 h研究中,我们主要关注神经元的PERK 通路。
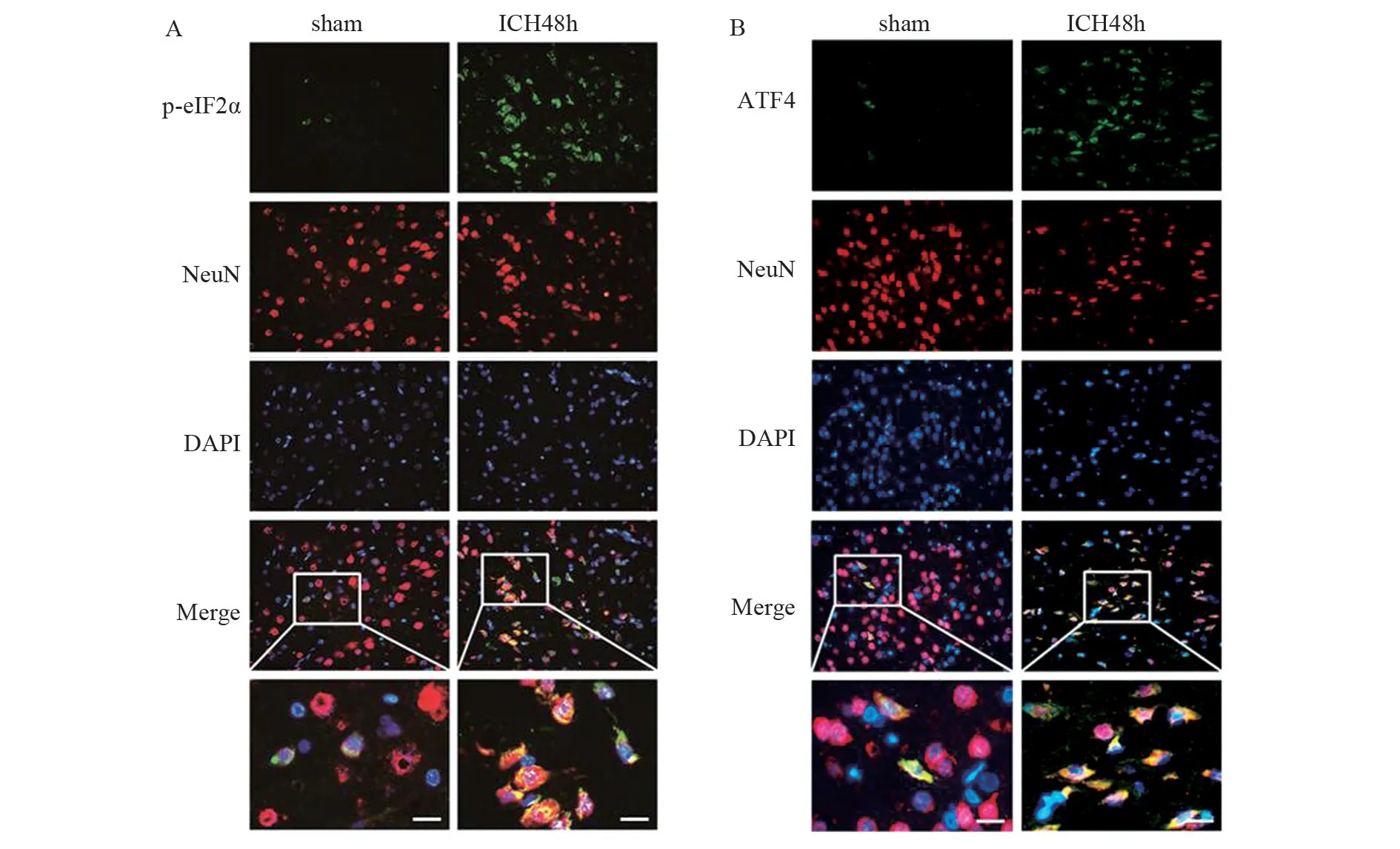
图3 免疫荧光双染分析脑组织中p-elF2α与ATF4的表达
2.4 PERK 通路促进ICH 诱导的体内凋亡
在ICH 后1 h 在脑室内注射PERK 抑制剂GSK2606414,在ICH 前30 min腹腔注射eIF2α去磷酸化抑制剂salubrinal,作为活跃的兴奋剂下游信号通路,实验结果如图4 所示。与ICH 组相比,GSK2606414组中p-eIF2α 和ATF-4 蛋白表达减少;与GSK2606414组相比,salubrinal组中p-eIF2α和ATF-4蛋白含量增加,见图4A。此外,在本研究中我们发现GSK2606414 处理可以显著逆转ICH诱导的CHOP和cleaved caspase-12蛋白水平的升高,见图4B。同时,根据神经细胞凋亡的组织学证据,ATF-4双标记TUNEL。结果显示:与sham 组相比,ICH 后TUNEL 和ATF-4 双阳性细胞数量增加,但GSK2606414处理可以消除了这种效应,见图4C。另外,salubrinal处理的效果与ICH 组相似,见图4C。上述结果提示,PERK 通路抑制可挽救ICH 诱导的神经元凋亡和坏死。

图4 PERK通路的抑制和激活对lCH导致的继发性脑损伤影响
2.5 PERK 信号通路促进ICH 诱导体外神经元凋亡
我们还进一步研究了PERK 信号通路在经Oxy Hb处理的初级神经元中的作用。与体内实验结果相似,与Oxy Hb组相比,GSK2606414组中peIF2α和ATF-4的蛋白质含量明显下降,与Oxy Hb组相比,salubrinal组中p-eIF2α和ATF-4 的蛋白质含量明显增加,见图5A。在Oxy Hb 处理下,CHOP和cleaved-caspase-12蛋白水平显著升高,提示诱导细胞凋亡,见图5B;与Oxy Hb 组相比,GSK2606414组 中CHOP 和cleaved-caspase-12 蛋白水平均显著降低,salubrinal组中CHOP 和cleaved-caspase-12蛋白水平均显著升高,见图5B。Tunel染色结果发现,与Oxy Hb组相比,GSK2606414 组中原代神经元数量减少了,salubrinal组中原代神经元数量增加了,见图5C。

图5 PERK通路的抑制和活化对氧合氢酶诱导的神经细胞体外凋亡的影响
3 讨论
蛋白激酶R 样内质网激酶(PERK)信号通路在神经元凋亡中发挥重要作用。脑出血会导致脑损伤,可破坏细胞代谢,激活细胞应激反应,包括UPR和ER 应激,ER功能的破坏导致ER应激[11]。氧化应激、线粒体钙超载、细胞离子平衡紊乱和有毒谷氨酸释放已被证明在各种疾病中诱导ER 应激,PERK 通路作为内质网应激的重要中介在神经元的命运中起着重要的作用,内质网应激诱导神经元凋亡;PERK/eIF2α信号已被证明在中枢神经系统损伤中调节神经元[12-13]。在本研究中,我们发现在脑出血后的神经元中,p-elF2α和ATF4 的蛋白水平明显升高,在48 h达到峰值,在体外和体内的结果是一致的。
PERK 信号通路作为内质网应激的中介,参与蛛网膜下腔出血后神经元凋亡;PERK 被AKT 相关的抗凋亡通路抑制,减少早期脑损伤[14];在创伤性脑损伤中PERK 和eIF2α的水平升高。内质网应激可导致细胞凋亡,抑制内质网应激可促进神经元存活,改善神经功能[15]。本研究发现:GSK2606414抑制p-eIF2α 和ATF4 的表达,通过抑制细胞的凋亡来促进神经元细胞的存活。此外,PERK 抑制剂降低了CHOP 和cleaved caspase-12的表达。因此,抑制PERK 信号在ICH 后具有神经保护作用。事实上,GSK2606414作为一种选择性PERK 抑制剂,在帕金森病和阿尔茨海默病中具有神经保护作用[16]。一些研究[17]表明,在各种疾病中,PERK 激活的增加可以减少神经元的凋亡。在中枢神经系统损伤后ER 和UPR 是利是弊尚未达成共识,而PERK 信号在ICH 后SBI中的作用尚不清楚。既往研究[18]表明,轻度刺激可激活ER 作为宿主防御机制,通过自噬使受损细胞器和蛋白降解,促进神经元存活。内质网应激不能对抗严重和长期的刺激,导致细胞凋亡。血肿周围水肿和ICH 后血肿的生理反应可引起SBI。
ICH 后,血液成分、功能失调的细胞器、过量产生的铁复合物和细胞因子水平持续升高,导致正常蛋白折叠被破坏,ER 和UPR 被激活,从而导致ICH 相关的脑损伤[19]。在人类中,颅内血肿在数周的时间内逐渐消退,在此期间大脑持续受到损伤,导致ER 时间延长,加重,最终导致神经细胞凋亡。同时,本研究也有一些局限性:首先,在本研究中,我们只专注于PERK 信号的作用成年雄性大鼠虽然ICH 可影响雌性,但在老年大鼠中很常见;其次,前期研究[20]表明PERK 通路通过直接相互作用促进钙调神经磷酸酶活性,胞质钙超载被认为是钙调神经磷酸酶诱导细胞凋亡的潜在机制。因此,PERK与钙调神经磷酸酶的确切关系值得在未来的研究中进一步研究。
综上所述,PERK 信号通路抑制可通过抑制细胞凋亡降低ICH 后SBI。基于这些发现,我们认为PERK 信号通路可能是神经元内源性生理调节信号通路的关键,可能是ICH 后减轻SBI的治疗靶点。
