健康成人口腔微生物组成及功能的宏基因组学研究
2022-04-29李玉姣程小刚钱飞潘雅婷陈力元田宇
人类口腔中生长着大量的细菌、真菌、病毒和古生菌等微生物,形成了一个丰富的微生物群落,被称为口腔微生物群,是人类体内仅次于结肠微生物群的第二复杂的微生物群落
。口腔微生物群与宿主微环境之间复杂的相互作用,维持着口腔微生态区域的动态平衡。口腔微生态一旦失衡,不仅会导致龋病
、牙周病
和口腔癌
等口腔疾病,还会引起糖尿病
、心血管疾病
、类风湿性关节炎
等全身系统性疾病。口腔微生物已经成为了探究口腔疾病及相关全身系统性疾病生物标志物的途径,深入了解健康人群口腔微生物组的物种组成及功能基因代谢通路十分必要。
该病是气传病害,必须采取以种植抗病品种为主,药剂防治和栽培措施为辅的综合防治策略,才能有效地控制其为害。
随着科技进步,人们对口腔微生物的研究不再局限于传统的培养方法,而是有了一系列的高通量测序技术。比如常用的16S ribosomal DNA(16S rDNA)扩增子测序技术和宏基因组学测序技术。两种方法相比较而言,16S rDNA 扩增子测序主要依赖于16S 基因中特定区域的测序,而选择的这种特定区域可能代表性不足,导致生物类群分类学分析分辨率有限
。而Handelsman 等
于1998 年提出了宏基因组的概念,是指生态环境中全部微小生物遗传信息的总和。与16S rDNA 扩增子测序不同,宏基因组测序具有较高的分辨率,可以直接获得可培养和不可培养微生物的全部遗传信息,不仅可以发现一些丰度较低的细菌类群,而且可以分析样本的功能基因,从而更深入地揭示口腔微生物群落的多样性和复杂性
,为研究口腔微生物提供了极大的便利。
那天的黄昏是记忆中最美的,在清澈流淌的小河边,一个简陋而又能遮风雨的棚,雨后的斜阳渗着无力的光,透过棚架,映在烟雾缭绕的方寸之地,生成了冷抽象的分割。昏暗中有人挂起了明亮的气灯,灯在风中摇曳,在动感的灯光与太阳的余晖交相掩映的瞬间,我看到了人们粗犷而善良的眼神,闪烁在惊喜的空间,那氛围使人突然迸发出一种让人毕生难忘的感动。
目前对健康成人口腔微生物群落功能基因的研究并不多,在对人类口腔微生物的研究中多采集唾液、龈上菌斑、龈下菌斑这三种口腔微环境样本,但由于本研究受试者的选择均为牙周健康者,可供龈下微生物生长的空间有限,因此本研究拟采用宏基因组学测序来描述健康成人受试者的唾液和龈上菌斑微生物群落组成及功能代谢途径,为今后实现口腔疾病的微生物学精准防治提供依据。
经空军军医大学第一附属医院医学伦理委员会批准(审批号:KY20212053-F-2),本研究选取社区人群中符合纳入排除标准的健康受试者共10 名,平均年龄(42.20 ± 11.27)岁,男5 例(平均年龄40.00 岁),女5 例(平均年龄44.40 岁)。纳入标准: ①年龄≥18 岁;②同意参与本实验并自愿签署知情同意书;③全口无明显活动性龋坏、牙周病、口腔脓肿、癌前病变、真菌感染和智齿冠周炎等口腔疾病;④所有受试者的余留牙数均大于等于28,龋失补指数(decayed,missing and filled teeth,DMFT)为0。排除标准: ①近6 个月内全身使用抗生素或免疫抑制剂等;②患有需要长期用药治疗的全身系统性疾病,如糖尿病、肿瘤、心脑血管疾病等;③艾滋病、乙肝、梅毒等传染性疾病;④孕期及哺乳期的妇女;⑤近6 个月内曾有过任何口腔药物及手术治疗史。所有研究对象均签署知情同意书。
1 资料和方法
1.1 研究对象的选择
阿里又使劲点点头。阿东又说:“罗爹爹去东湖练拳,走不动了。我们阿里每天早上用爸爸的轮椅推罗爹爹去,好不好?”
本组70例病患都接受CT和磁共振检查,详细如下:(1)CT检查。指导取仰卧位,选择双层螺旋CT扫描仪,并设置层间距10~15mm,层厚20mm,然后再对病灶进行全面的扫描,同时选择病灶5mm厚度的部位进行加层扫描。(2)磁共振检查。指导取仰卧位,利用体线圈,便于成像。利用磁共振仪对患者施以全面的扫描,设计层厚为4~9mm,然后再对病灶施以4mm的薄层扫描。
1.2 样本采集
所有受试者采样前1 天晚饭后刷牙,然后禁饮食、禁止刷牙,直至第二天早上进行唾液和龈上菌斑样本的采集。唾液样本:采集样本之前嘱咐受试者切勿咳痰,采用非刺激性的方法获得唾液样本,让唾液自然流入50 mL 无菌离心管中,收集量至少达到10 mL(唾液中不含血、食物残渣、痰等杂质)。龈上菌斑样本:使用无菌刮匙刮取受试者上下颌前牙、前磨牙、磨牙颊舌侧牙颈部菌斑,并置于含有PBS 缓冲液的1.5 mL 无菌EP 管中。对所有采集到的样本进行分组编号(唾液组命名为A,龈上菌斑组命名为B,共20 个样本)后置于液氮里冷冻,并在2 h 内送至实验室,放于-80 ℃冰箱内保存。
1.3 样本DNA 提取
根据产品制造商的说明,通过NEBNext 微生物组DNA 富集试剂盒(New England Biolabs,美国)提取唾液及龈上菌斑样本微生物群落DNA。使用Qubit dsDNA BR 分析试剂盒(Invitgen,美国)和荧光光度计(Qubit Fluorometer,美国)对微生物群落DNA 进行定量,并在1%琼脂糖凝胶上进行质量检查。
定期邀请已毕业的优秀学生和公司的人力资源主管给学生作报告,由他们讲述公司的人才需求和评价标准,对学生在校学习也有积极作用。每年学校的毕业生双选会,食品科学与工程专业都会邀请行业知名企业高管参加座谈会,把脉行业发展趋势,探讨行业企业对毕业生的知识能力素质需求,站在企业的角度上探讨学生评价的标准,改进现有的教学评价系统,促进学生全面发展。
1.4 宏基因组文库的构建
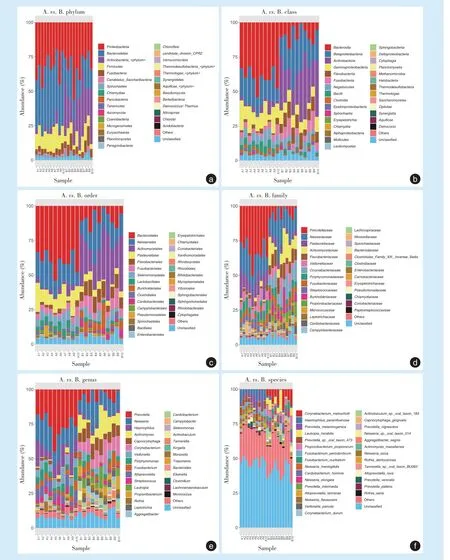
本实验中20 例样本共注释到49 个门、72 个纲、146 个目、322 个科、1 185 个属、4 450 个种,对各个物种分类水平上的菌群分布结果进行分析,详见图2。在门水平,所有样本的共有优势菌门为变形菌门(32.51%)、拟杆菌门(30.81%)、放线菌门(16.23%)、厚壁菌门(10.49%)、梭杆菌门(5.45%)、
门(1.03%)、螺旋体门(0.48%)、衣原体门(0.15%)、浮霉菌门(0.05%)、蓝细菌门(0.04%),这10 个菌门占据了97.24% 以上。在种水平,所有样本的共有优势菌种为马氏棒状杆菌(3.84%)、副流感嗜血菌(2.91%)、产黑色素普雷沃菌(2.76%)、奇异劳特普罗菌(2.47%)、
(2.01%)、丙酸丙酸杆菌(1.99%)、牙周梭杆菌(1.44%)、具核梭杆菌(1.19%)、脑膜炎奈瑟菌(1.13%)、人心杆菌(1.12%),共占了20.86%以上,约44.21%的菌种尚未被命名,表明了人类口腔微生物群落中仍有大量物种尚未被发现。
1.5 上机测序及数据组装
质控合格的文库在MGISEQ-2000 平台(BGI-深圳,中国)测序。所有原始数据用软件SOAPnuke(v.1.5.2)进行修剪。为了获得功能信息,将蛋白质序列与eggNOG 数据库(2015-10),CAZy 数据库(2017-09),KEGG 数据库(89.1)等进行功能注释,E值的截止值为1e
。
1.6 生物信息学分析和统计学分析
通过长久的相互适应及功能融合,口腔微生物群落已经和人体组成了一个完整的有机体。口腔微生物群落失调往往会导致疾病的发生,应深入了解、探究健康状况下的口腔微生物发生发展规律,从而设法维持口腔微生态环境的平衡,保持机体健康。近年来对口腔微生物的研究方法有培养法、16s rDNA 测序及宏基因组学测序方法。因为许多种类口腔微生物区系是不可培养的,故传统培养法限制了对口腔微生物群的研究;16s rDNA 测序方法虽然能发掘无法培养的细菌、培养阴性的细菌、稀有细菌等
,但是它并不能在种水平上提供足够高的分辨率来识别细菌,且不能同时探究口腔微生物的重要非细菌成分,包括真核微生物(真菌、原生动物)和病毒等
。因此,本研究采用具有高分辨率的宏基因组学方法对健康成人的口腔微生物进行物种组成及功能分析。
2 结 果
2.1 测序数据统计
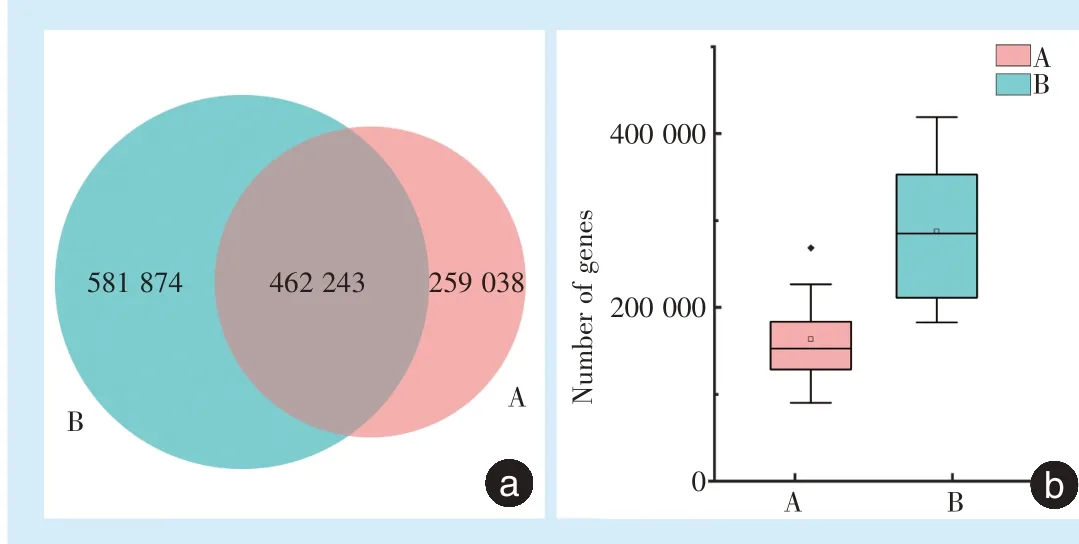
构建唾液及龈上菌斑基因集的维恩图(图4a),两组基因集中包含共有基因462 243 个,唾液样本中特有基因有259 038 个,龈上菌斑样本组特有基因有581 874 个。龈上菌斑组基因集中的基因数目远高于唾液组(图4b)。
2.2 物种稀疏曲线分析
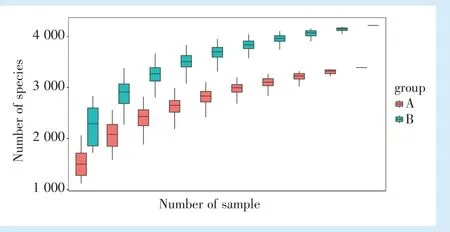
为了检验本实验样本量是否足够,对唾液和龈上菌斑样本的测序物种数目进行稀疏曲线分析(唾液组命名为A,龈上菌斑组命名为B),如图1 所示:随着样本量的增加,稀疏曲线末端逐渐平缓,检测到的物种数目不再增加,表明样本量能够满足本实验的要求。
2.3 健康口腔样本在各分类水平上的微生物组成
用Covaris 仪(Covaris,美国)随机扩增1 μg 质检合格的DNA。用磁珠筛选片段DNA,平均大小为200~400 bp。筛选出的片段经过末端修复,3’端腺苷酸、接头连接、PCR 扩增,产物经磁珠纯化。对双链PCR 产物进行热变性,并用夹板寡核苷酸序列进行环化。将单链环状DNA 格式化为最终文库,并经质控鉴定。
2.4 物种多样性分析
2.4.1 唾液和龈上菌斑两组间Alpha 多样性分析对唾液样本及龈上菌斑样本微生物群落在种水平上的Alpha 多样性(Shannon、Simpson、Chao1 指数)进行分析,详见表1。龈上菌斑组的Alpha 多样性指数均高于唾液组。通过Wilcoxon 秩和检验分析,两组Alpha 多样性指数差异均有统计学意义(
<0.05),说明龈上菌斑样本的物种丰富度和均匀度均高于唾液样本。

利用Wilcoxon 非参数秩和检验进行唾液和龈上菌斑两群落间种水平上的物种组成差异分析。结果显示:唾液微生物中副流感嗜血菌、产黑色素普雷沃菌、牙周梭杆菌、中间普雷沃菌、坦纳拟普雷沃菌、浅黄奈瑟菌、变黑普雷沃菌、组织普雷沃菌、轻型链球菌等较龈上菌斑丰富(
<0.05);而龈上菌斑样本中马氏棒状杆菌、产酸丙酸杆菌、黏性放线菌、奇异劳特普罗菌、丙酸丙酸杆菌、具核梭杆菌、延长奈瑟菌、内氏放线菌、牙龈二氧化碳噬纤维菌、马氏放线菌、龋齿罗氏菌等比唾液中丰富(
<0.05)。
2.5 唾液和龈上菌斑两群落间种水平上的物种组成差异
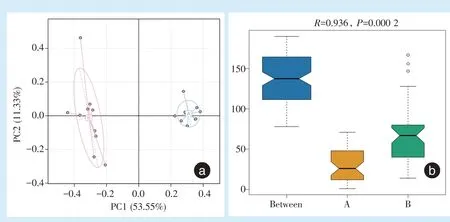
2.4.2 唾液和龈上菌斑两组间微生物群落Beta 多样性分析 唾液与龈上菌斑两组样本微生物群落种水平上的Beta 多样性采用主成分分析(principal components analysis,PCA)和Anoism 分 析 进 行 计算。结果如图3a 和图3b 所示:PCA 分析表明两组样本间明显能各自聚类,Anoism 分析表明唾液与龈上菌斑样本的微生物群落构成差异有统计学意义(
=0.936,
=0.000 2)。
2.6 健康成人唾液及龈上菌斑基因集构建
20 个样本测序下机数据量(raw data)均值为11 817 225 240,去除低质量数据,获得高质量数据(clean deta)量均值为11 421 498 690,有效数据率均值为96.74%。
2.7 唾液与龈上菌斑微生物功能组成及代谢通路差异
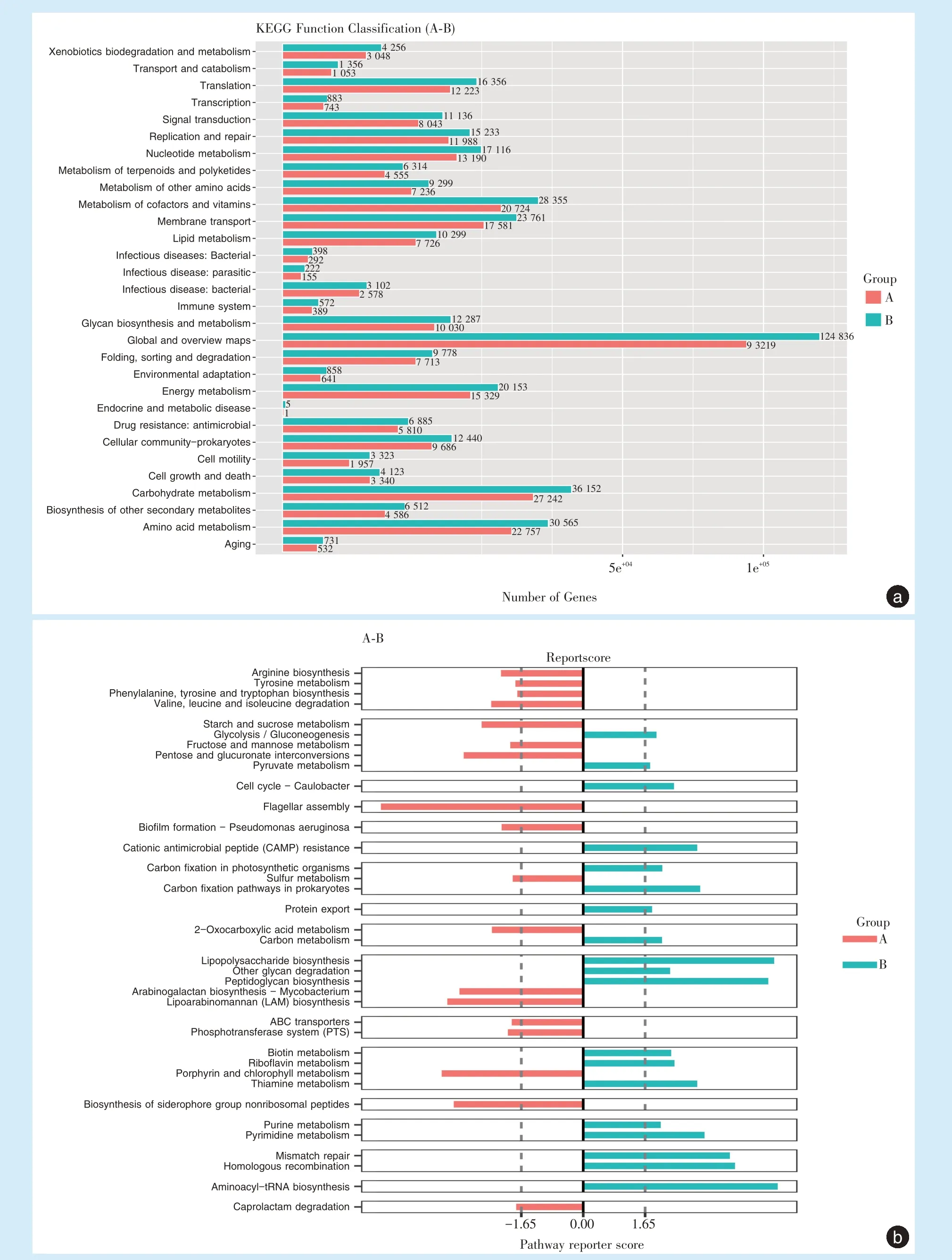
KEGG Orthology 功能数据库注释得到唾液和龈上菌斑微生物功能代谢途径的直观分布图,如图5a 所示,口腔微生物KEGG 功能代谢途径主要集中在碳水化合物代谢、氨基酸代谢、辅酶和维生素的代谢、膜转运、能量代谢。利用KEGG 通路数据库对唾液和龈上菌斑微生物的功能代谢途径差异进行了鉴定,得到两组间共有53 个差异代谢通路(reporter score 绝对值>1.65),按丰度值取前38 名作功能代谢通路差异分布图,结果如图5b 所示。结果显示唾液微生物功能代谢途径中鞭毛组装、铁载体非核糖体肽的生物合成、戊糖-葡萄糖醛酸转换、淀粉与蔗糖的代谢、亮氨酸和异亮氨酸降解、精氨酸生物合成较龈上菌斑丰富,而龈上菌斑中丙酮酸代谢、糖酵解/糖合成、嘌呤代谢、碳代谢、生物素代谢、核黄素代谢等途径较唾液中丰富。
3 讨 论
利用R 语言包在物种、功能基因和KO 水平上进行组成分析,用Shannon 指数、Simpson 指数、Chao1 指数对微生物α 多样性进行量化,基于样本相似性距离矩阵进行PCA 分析及Anoism 分析计算β 多样性;使用SPSS 26.0 软件进行统计学分析,通过Wilcoxon 非参数检验确定不同组间物种及功能差异显著的特征(
<0.05);采用MetaGeneMark 算法程序对测序得到的宏基因组基因从头预测,然后通过CD-hit 算法程序进行基因集去冗余得到高质量基因集。根据Reporterscore 评分值确定差异富集的KEGG 通路,以评分绝对值≥1.65 作为显著性检测阈值。
充足的样本量是研究的基础和前提,能够保证研究信息的完整性。本研究通过稀疏性曲线分析结果可知,随着样本数目增加,曲线末端趋于平缓,说明本实验样本数量是足够的。通过物种注释得到达1 185 个属、4 450 个种,检测到的各分类学水平的物种数量远远超过与同类研究16S rDNA口腔微生物测序的结果
。微生物组成分析结果显示口腔微生物中变形菌门、拟杆菌门、放线菌门、厚壁菌门、梭杆菌门等占整个口腔微生物群菌门的97.2%,与现有人类口腔微生物基因组数据库的结果一致
。这些研究表明在不相关的健康个体中发现的大多数口腔分类群是相似的,并且支持了口腔健康核心微生物组的概念。
口腔是一个非常复杂的环境,不同的微生物优先在不同的栖息地定居
。深入分析掌握口腔各空间不同微生态的菌群特点,可能是人类成功调节口腔微生物及进行口腔疾病防治的关键。本研究结果显示,唾液和龈上菌斑样本微生物群落微生物多样性及菌群构成具有明显差异,龈上菌斑相比唾液微生物具有更高的丰富度和均匀度,唾液微生物中产黑色素普雷沃菌、牙周梭杆菌、中间普雷沃菌、变黑普雷沃菌等牙周致病菌较龈上菌斑丰富,而龈上菌斑样本中产酸丙酸杆菌、黏性放线菌、丙酸丙酸杆菌、具核梭杆菌、内氏放线菌、龋齿罗氏菌等龋病致病菌比唾液中丰富。由此笔者推测唾液微生物组特征对识别牙周病易感个体更加敏感,而龈上菌斑微生物组特征可能对识别龋病易感个体更加敏感;这一结果和最近几项研究相符,Yang 等
通过对乳磨牙和上颌第一恒磨牙的龈上菌斑和非刺激性唾液样本测序分析,发现相比唾液,龈上菌斑是研究龋病菌群的最佳微生物群落。此外,Acharya 等
利用唾液微生物种类将慢性牙周炎患者与健康对照组区分开来,并发现唾液微生物组特征或许能够准确识别出牙周炎易感个体。但是由于本实验样本量有限且口腔微生物受到研究对象饮食、生活习惯等多种因素的影响,这就限制了以上结果的普适性。本研究结果显示物种丰度分布在两个细菌群落之间有很大的不同,Segata 等
研究结果也表明龈上菌斑微生物中棒状杆菌属、二氧化碳噬纤维菌属、罗氏菌属、卟啉单胞菌属相对丰度占据了2%以上,而唾液微生物中,链球菌属、韦荣菌属、普雷沃菌属、奈瑟菌属、梭杆菌属、放线菌属和纤毛菌属的相对丰度均超过2%。龈上菌斑和唾液微生物的这种显著差异可能来源于多种因素,不同口腔微生态环境的理化性质不同,唾液微生物多为浮游微生物,而牙齿表面为龈上菌斑提供了不同的细菌粘附受体,生物膜内部的协同和拮抗作用也可能会介导菌群不同的定植和分布
。
不同口腔微生态菌群的功能代谢途径也可能不同。本研究发现口腔健康受试者的口腔微生物KEGG 功能代谢途径主要集中在碳水化合物代谢、氨基酸代谢、辅酶和维生素的代谢等。其中,碳水化合物代谢是口腔微生物的重要代谢途径。当碳水化合物途径发生变化时,口腔微生态的生态平衡便会被打破,口腔致病菌也会随之变化
,比如糖酵解途径增加,会使口腔微生态呈现酸性环境,变形链球菌、乳酸杆菌等耐酸菌便会成为优势菌种,并最终导致龋病的发生
。本研究唾液和龈上菌斑微生物功能代谢途径差异分析结果显示,唾液中亮氨酸代谢较龈上菌斑丰富。亮氨酸是支链氨基酸,可以通过脱氨、脱羧、还原生成乙酰辅酶A,分别转化为异丁酸和异戊酸,而唾液中较丰富的中间普雷沃菌便可利用此途径
,这与本研究物种分布结果相符。同时唾液中的牙周梭杆菌和普雷沃特氏菌可以利用精氨酸,通过精氨酸脱亚胺酶及胍丁胺脱亚胺酶系统代谢途径中和酸性环境,生态环境一旦失衡可能促进更多这种牙周病细菌的引入
。龈上菌斑中丙酮酸代谢较唾液丰富,其中与之相对应的溶糖细菌如丙酸杆菌、放线菌可通过丙酮酸代谢途径,通过各种分支途径进一步转化为乳酸、醋酸、乙醇和甲酸盐
。以上结果均说明了对口腔微生物的研究不仅要关注微生物组成的改变,对口腔微生态功能基因代谢途径的探究能使人们对口腔疾病的发病机制更加了解。
综上所述,本研究对健康成人口腔微生物各分类学水平组成及功能基因进行了分析,发现产黑普雷沃菌、副流感嗜血菌、
在健康人群唾液内为优势菌群,马氏棒状杆菌、奇异劳特普罗菌、丙酸丙酸杆菌在健康人群龈上菌斑内为优势菌群,碳水化合物代谢在健康受试者口腔微生物代谢中起着重要作用。本研究结果可能和所有关注口腔微生物组及口腔疾病微生物变异的研究相关,可为描述各类型疾病期间发生的口腔微生物的失调状况提供基线理论参考。但本研究也存在一定的局限性,如样本数量有限导致研究结果可能不够全面;此外,本研究只对健康成人口腔微生物进行了分析,没有对牙周病、龋病等口腔疾病状态下的口腔微生物进行研究。因此,今后的研究还需扩大样本量对本实验结果进行验证,并对牙周病、龋病等口腔疾病状态下口腔微生物组成和功能基因的改变进行探究,以深入了解人类口腔微生物群落在健康和疾病中发挥的实际作用。
四川盆地长宁-威远页岩气开发示范区生产废水管理…………………………………………………………(4):113
Li YJ collected, measured and analyzed the data and wrote the article. Cheng XG analyzed the data. Qian F, Pan YT and Chen LY collected and measured the data. Tian Y designed the study and revised the article. All authors read and approved the final manuscript as submitted.
[1] Mosaddad SA, Tahmasebi E, Yazdanian A, et al. Oral microbial biofilms: an update[J]. Eur J Clin Microbiol Infect Dis, 2019, 38(11):2005-2019.doi:10.1007/s10096-019-03641-9.
[2] Jiang Q, Liu J, Chen L, et al. The oral microbiome in the elderly with dental caries and health[J].Front Cell Infect Microbiol, 2018,8:442.doi:10.3389/fcimb.2018.00442.
[3] Willis JR, Gabaldón T. The human oral microbiome in health and disease:from sequences to ecosystems[J].Microorganisms, 2020,8(2):308.doi:10.3390/microorganisms8020308.
[4] Chen WP, Chang SH, Tang CY, et al. Composition analysis and feature selection of the oral microbiota associated with periodontal disease[J].Biomed Res Int,2018(1):3130607.doi:10.1155/2018/3130607.
[5] Mok SF, Karuthan C, Cheah YK, et al. The oral microbiome community variations associated with normal, potentially malignant disorders and malignant lesions of the oral cavity[J]. Malays J Pathol,2017,39(1):1-15..
[6] Long J, Cai Q, Steinwandel M, et al. Association of oral microbiome with type 2 diabetes risk[J]. J Periodontal Res, 2017, 52(3):636-643.doi:10.1111/jre.12432.
[7] Farrugia C, Stafford GP, Murdoch C.
outer membrane vesicles increase vascular permeability[J]. J Dent Res,2020,99(13):1494-1501.doi:10.1177/0022034520943187.
[8] Liu X, Tian K, Ma X, et al. Analysis of subgingival microbiome of periodontal disease and rheumatoid arthritis in Chinese: a casecontrol study[J]. Saudi J Biol Sci, 2020, 27(7): 1835-1842. doi:10.1016/j.sjbs.2020.04.040.
[9] Laudadio I, Fulci V, Palone F, et al. Quantitative assessment of shotgun metagenomics and 16S rDNA amplicon sequencing in the study of human gut microbiome[J]. OMICS, 2018, 22(4): 248-254.doi:10.1089/omi.2018.0013.
[10] Handelsman J, Rondon MR, Brady SF, et al. Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products[J]. Chem Biol, 1998, 5(98): 90108-90109.doi:10.1016/s1074-5521(98)90108-9.
[11] Durazzi F, Sala C, Castellani G, et al. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota[J]. Sci Rep, 2021, 11(1): 3030. doi:10.1038/s41598-021-82726-y.
[12] Woo PC, Lau SK, Teng JL, et al. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories[J].Clin Microbiol Infect, 2008, 14(10): 908-934. doi: 10.1111/j.1469-0691.2008.02070.x.
[13] Caselli E, Fabbri C, D'accolti M, et al. Defining the oral microbiome by whole-genome sequencing and resistome analysis:the complexity of the healthy picture[J].BMC Microbiol, 2020,20(1):120.doi:10.1186/s12866-020-01801-y.
[14] 李宗泽,赵泽,张文菲,等.16S核糖体DNA Amplicon 测序法分析2型糖尿病患者口腔微生物多样性[J].转化医学杂志,2017,6(3):151-156.doi:10.3969/j.issn.2095-3097.2017.03.006.Li ZZ,Zhao Z,Zhang WF,et al.Analysis of oral microbial diversity in patients with type 2 diabetes mellitus by 16S ribosome DNA Amplicon sequencing[J].Translational Med J, 2017,6(3):151-156.doi:10.3969/j.issn.2095-3097.2017.03.006.
[15] Verma D,Garg PK,Dubey AK.Insights into the human oral microbiome[J]. Arch Microbiol, 2018, 200(4): 525-540. doi: 10.1007/s00203-018-1505-3.
[16] Yang X,He L,Yan S,et al.The impact of caries status on supragingival plaque and salivary microbiome in children with mixed dentition: a cross-sectional survey[J]. BMC Oral Health, 2021, 21(1):319.doi:10.1186/s12903-021-01683-0.
[17] Acharya A,Chen T,Chan Y,et al.Species-Level salivary microbial indicators of well-resolved periodontitis:a preliminary investigation[J]. Front Cell Infect Microbiol, 2019, 9: 347. doi: 10.3389/fcimb.2019.00347.
[18] Segata N,Haake SK,Mannon P,et al.Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces,tonsils, throat and stool samples[J]. Genome Biol, 2012, 13(6):R42.doi:10.1186/gb-2012-13-6-r42.
[19] Kaan A, Kahharova D, Zaura E. Acquisition and establishment of the oral microbiota[J]. Periodontol 2000, 2021, 86(1): 123-141.doi:10.1111/prd.12366.
[20] Takahashi N. Oral microbiome metabolism: from "who are they?"to "what are they doing?"[J]. J Dent Res, 2015, 94(12): 1628-1637.doi:10.1177/0022034515606045.
[21] Tanner ACR, Kressirer CA, Rothmiller S, et al. The caries microbiome: implications for reversing dysbiosis[J]. Adv Dent Res,2018,29(1):78-85.doi:10.1177/0022034517736496.
[22] Zhang Y,Zhen M,Zhan Y,et al.Population-Genomic insights into variation in prevotella intermedia and prevotella nigrescens isolates and its association with periodontal disease[J]. Front Cell Infect Microbiol,2017,7:409.doi:10.3389/fcimb.2017.00409.
[23] Huang X,Zhang K,Deng M,et al.Effect of arginine on the growth and biofilm formation of oral bacteria[J]. Arch Oral Biol, 2017, 82(82):256-262.doi:10.1016/j.archoralbio.2017.06.026.
[24] De Oliveira R, Bonafé F, Spolidorio D, et al.
and
interaction in dual-species biofilm[J].Microorganisms, 2020, 8(2): 194. doi: 10.3390/microorganisms80 20194.
