基于量子化学的药物共晶虚拟筛选方法研究进展*
2022-04-27于铭超王志鹏杨世颖杨德智吕扬
于铭超,王志鹏,杨世颖,杨德智,吕扬
(北京协和医学院、中国医学科学院药物研究所晶型药物研究北京市重点实验室,北京 100050)
药物共晶作为一种新兴的前沿技术,可以在不改变药物活性成分(active pharmaceutical ingredient,API)共价结构[1]的前提下,定向改善药物的理化性质[2]、降低毒副作用[3]、增强药效活性[4]。此外,参与共晶形成的共晶形成物(cocrystal former,CCF)的多样性在一定程度上扩大了共晶的种类和数量[5],在延长API专利保护期的同时,创造了新的商业价值[6]。这些独特的优势,使其在近年来受到学术界、产业界和药品监督管理部门的广泛关注。随着人们对共晶研究兴趣的不断增加,高通量筛选药物共晶的实验技术随之迅速发展[7],例如基于溶液生长的筛选方法[8]、溶剂滴磨法[9]、成浆[10]及实验测定相图法[11]等。然而,这些以实验为基础的共晶筛选方法普遍存在耗时长、操作复杂、成本昂贵以及污染环境的缺点,无法适应药物研究开发与社会发展的需要。随着多学科的交叉发展,量子化学融合并应用于分子结构研究中,在分析共晶物的组成、构象和分子内/分子间相互作用等方面发挥重要作用[12-13]。因此,基于量子化学理论的虚拟筛选技术应运而生,例如基于球形原子电子密度之和的Hirshfeld表面分析,被认为是研究共晶物所表现出的非共价相互作用的有效工具[14-15];基于密度泛函理论的分子表面静电势可以用来识别分子表面上可能的分子间相互作用位点[16];分子中原子的量子理论计算提供了包含氢键在内的大量信息,键临界点有助于识别化学键和原子间相互作用,这些拓扑性质有助于表征氢键的强度和性质[17-19];Hansen溶解度参数( Hansen solubility parameter,HSP)描述了分子通过分子间作用力的可能性,成为预测共晶形成的常用工具[20];基于液相热力学理论的COSMO-RS(conductor-like screening model for real solvents) 计算模型由于其考虑了氢键等分子相互作用[21],也广泛用于药物共晶的高通量筛选。随着理论计算的发展和计算机技术的不断进步,共晶虚拟筛查技术正在不断完善。
笔者综述3种常用的基于量子化学计算方法的虚拟共晶筛选方法的原理及其在药物共晶的预测与筛选方面的作用,旨在通过介绍3种方法的原理、适用范围及优缺点,为其在药物共晶虚拟筛选方面的合理应用提供参考。
1 基于分子表面静电势(molecular electrostatic potentials,MEPs)的虚拟共晶设计方法
1.1MEPs MEPs通常是指依据Bader定义的电子密度为0.001 e/Bohr^3的等值面[22],根据不同区域静电势大小通过不同颜色进行绘制,从而得到可以三维描述分子表面电趋的图谱。长时间以来,MEPs一直被用作分析和阐释小分子化合物[23]及大分子络合物[24]等多种体系的各种结构特征[25]和非共价相互作用[26]。20世纪80年代开始,MEPs在药物设计领域开始崭露头角[27],但是直到最近,MEPs对超分子相互作用的识别才广泛应用于晶体工程领域[28-32]。
对于大多数共晶来说,氢键在API与CCF的识别与连接中发挥重要作用[33-37],因此可以以氢键为基础来设计和筛选药物共晶。根据氢键形成规律[38],MUSUMECI等[16]提出了虚拟共晶设计理论,通过MEPs计算识别分子表面上可能的分子间相互作用位点。
假设API和CCF分子表面所有可能相互作用位点都可以彼此相互作用,通过对所有相互作用位点配对能进行简单求和来评估这些分子的总相互作用位点配对能量(E)。比较两种纯组分和共晶之间的能量差异ΔE是评估共晶形成可能性的一个重要策略。

(1)

(2)
式中,MEPmax和 MEPmin分别为 MEPs 上的局部最大值和最小值,单位kJ·mol-1。
E=- ∑ αiβj
(3)
式中,αi为氢键供体参数;βj为氢键受体参数;E为这些分子的总相互作用位点配对能量。
ΔE=Ecc-nE1-mE2
(4)
式中,E1为化合物1纯态相互作用位点配对能;E2为化合物2纯态相互作用位点配对能;Ecc为化学剂量比为1n2m的共晶相互作用位点配对能。
根据用氢键参数αi和βj乘积估算的化合物分子相互作用位点配对能,从而对氢键供体位点和受体位点进行等级划分,最佳的氢键供体与最佳的氢键受体配对、次佳的氢键供体与次佳的氢键受体配对,以此类推,这为分子间识别提供了可靠的指导原则。
MUSUMECI等[16]首先利用Spartan '06软件对化合物MEPs进行计算,得到MEPs局部最大值和最小值。近年来,更多科研工作者选择功能强大的Gaussian程序先优化分子构象,再利用免费的Multiwfn程序从MEPs提取局部极大值和极小值来进行计算[39-41]。获得MEPs局部最大值和最小值后,使用公式(1)、(2)将MEPs转换成相应的氢键参数α和β,然后利用公式(3)用氢键参数α和β的乘积来近似表示相互作用位点总配对能量,之后根据公式(4)计算纯组分与共晶的相互作用配对能的差值,-ΔE越大,其共晶的形成率越高,从而估算两个化合物形成共晶的概率。
1.2MEPs在药物共晶的预测与筛选中应用与发展 MEPs的应用实际上是假设固体优化了所有可能的相互作用[16],即分子表面上的所有氢键位点都是独立的,相邻的氢键位点不会相互影响,并且可以自由地在固体中找到它们的最佳配对位点。忽略晶体中分子的三维结构和空间排列,即接触没有空间限制,没有堆积效应,分子表面空间相近的位点之间没有协同性。这种方法比较粗糙,涉及了许多假设,并且基于主要的近似值,但是它的优点是应用起来十分快速、简便,可以对备选共晶配体进行大规模、快速地筛选。
近年来,MEPs理论计算在共晶形成的预测作用得到了广泛的应用,例如ZHANG等[29]通过对黄酮类化合物和4,4′-乙烯基二吡啶的MEPs进行计算,预测了共晶中可能存在的氢键杂合子以及API与共晶配体的摩尔比,在此基础上,成功合成了10种4,4′-乙烯基二吡啶-黄酮类化合物共晶的单晶。GRECU等[42]对18种药物活性成分进行MEPs分析计算,根据ΔE的值对共晶配体进行排序筛选,筛选结果与API的实验性共晶筛选文献数据基本一致,验证了MEPs理论计算在共晶配体筛选过程中的可行性。
然而,值得注意的是,基于MEPs的虚拟计算虽然计算简单、耗时短,但是计算过程忽略了分子构象对MEPs上极值点分布的影响,计算结果的偏差会对预测晶体形成的成功率产生影响。鉴于在影响共晶分子间相互作用的因素中,分子构象至关重要,YANG等[30]将构象分析引入到MEPs分析中,选取化合物热力学稳定的主要构象开展研究,并结合化合物氢键供受体的官能团数量优化了相互作用配对能的算法,使得化合物产生相互作用后体系能量与各组成单体成分能量和的差值ΔE更能体现药物与配体间相互作用的强弱。该法针对于以静电相互作用为主导的1:1共晶的形成,预测成功率接近80%。该研究为提高MEPs共晶预测的成功率提供了新思路。
2 依据HSP的共晶设计方法
2.1HSP 溶解度参数(δ)的概念由HILDEBRAND等[43]首次提出,他们认为具有相同δ的溶剂是可互溶的。1967年,HANSEN等[44]提出了HSP概念,HSP模型将总内能(Et)分解为色散能(Ed)、极化能(Ep)和氢键能(Eh)。
Et=Ed+Ep+Eh
(5)
通过将方程两边除以摩尔体积V即可得到总溶解度参数(δt),也称为三维溶解度参数,可定义如下:

(6)

(7)
(8)

(9)
其中,i是分子内的结构基团,Fdi是基团对色散力的贡献,Fpi是基团对极性力的贡献,Fhi是基团对氢键能的贡献,Vi是基团对摩尔体积的贡献。
化合物分子的相溶性是由各种标准来估计的,它们都是基于“相似相溶”的原理,即具有相似δ值的化合物可能是可混溶的,δ值越接近,它们的相似性就越大,亲和力就越强,这种理论分析适用于聚合物溶解也适用于药物共晶领域[20]。

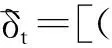
(10)
此外,BAGLEY等[49]注意到δd和δp在热力学上是相似的,而δh与两者的性质不同,因此引入了体积相关的溶解度参数δv,并提出了另一个标准,用Ra(v)因子测定了两种化合物的相溶性:

(11)
Ra(v)=[4(δv2-δv1)2+(δh2-δh1)2]0.5
(12)
在后来的研究中,GREENHALGH等[50]利用API与CCF总溶解度参数的差异(Δδt)作为预测混溶性的工具:
Δδt=|δAPI-δCCF|
(13)

汉森溶解度参数由ICAS程序计算[51],基于Gani的基团贡献法的20.1版本[52]。
2.2HSP在预测共晶的形成和筛选中的应用 HSP是一种简单的理论方法,只需要了解分子的化学结构[53]。鉴于HSP的独特优势,许多科研工作者[54-56]报道使用HSP作为共晶筛选方法的计算结果。GRECU等[57]甚至认为它是比晶格能第一原理计算更好的高通量筛选方法,然而GADADE等[58]则认为HSP是一种有限使用的筛选方法。
HSP在共晶筛选方面是一种很有前途的方法,只要使用一系列CCF与API进行混溶计算验证,并研究其他制备方法和条件,以确定混溶性是共结晶的先决条件[60],然而在该方法成为药物共晶筛选的常规方法之前,临界值需要重新评估和细化。
3 以液相热力学理论COSMO-RS计算模型为基础的共晶设计方法
3.1COSMO-RS COSMO-RS 是通过对量子化学COSMO溶剂化模型的改进,结合统计力学方法延伸出来的一种定量计算溶剂化现象的新方法[61],通过量子化学连续溶剂化计算,对极性键和氢键的相互作用能进行了量化[62]。改善了COSMO溶剂化模型原有的一些缺陷[63],广泛用于液相系统的热力学平衡性质的预测[64-65]。由于COSMO-RS理论考虑分子重要的相互作用,如氢键、范德华力和静电相互作用等[21],因而COSMO-RS可用于实现药物共晶的快速、高通量筛选。
COSMO-RS是一个液相热力学模型,因此可以计算共晶组分的虚拟过冷液体的相溶性,并获得由纯组分A和B产生的化学计量m:n混合物的过量焓(Hex)。
Hex=HAB-xmHpureA-xnHpureB
(14)
Hpure和HAB分别代表纯物相和m:n混合物中的摩尔焓,摩尔分数xm=m/(m+n)和xn= n/(m+n)。Hex是一个优于纯氢键相互作用的描述符,Hex包含的焓贡献,并不局限于氢键相互作用,这些可以通过COSMOtherm软件从总焓中分离出来[63]。在COSMO-RS计算中,API和CCF数据可直接从BP-TZVP-COSMO化合物数据库导出,然后导入COSMOthermXver2019以获得混合物的ΔHex[66]。
ABRAMOV 等[62]经过大量的实验计算,证明了Hex<0的化合物在溶液(过冷液体)中具有很强的相互作用,在焓值上更倾向于混合物而不是其纯液体。Hex可以解释为两种组分在共结晶之前在混合物中结合的趋势,Hex越负,这两个组分就越有可能形成多组分晶体,如共晶或盐[62,67]。此外,ABRAMOV 等[62]还证明了COSMOtherm允许对共晶配体进行合理的排序,以提高API的溶解度。与确定共晶的潜在共晶配体类似,形成固体溶剂化物的可能性最低的溶剂可根据API的最高Hex确定。此类溶剂体系可直接用于稳定形式的浆液结晶,以及通过重结晶实验实现溶剂化物的去溶剂化,以促进溶剂介导的转化和转化为稳定的无水非溶剂化物。
3.2COSMO-RS在药物共晶预测与筛选中的应用 尽管COSMO-RS也包含不可忽略的参数,但它们是元素特异性的,不受限于官能团,在使用前不需要任何重新参数化。COSMO-RS理论及其软件实现的独特预测能力[68]使其在药物共晶筛选研究中发挥重要的作用。
ROCA-PAIXAO等[67]应用COSMO-RS方法对API卡马西平和75种备选共晶配体进行了亲和力预测计算。将计算结果与实验结果进行了直接比较,对COSMO-RS方法用于缩短共晶设计的可能性和局限性进行了评估。结果表明,COSMO-RS理论在COSMOTherX软件中的实现为通过计算筛选预选共晶共形物提供了一种高效的方法。CHENNURU等[69]运用COSMO-RS理论及其软件成功预测并筛选了4种新新型乙酸阿比特龙酯共晶,极大地优化了乙酸阿比特龙酯自身的溶解性。LIU等[70]运用COSMOTherX 19程序对难溶性营养物乳清酸和41种备选共晶配体进行亲和预测,在亲和力预测和实验结果的基础上,选择甘氨酸、丙氨酸、4-氨基丁酸、5-氨基戊酸和6-氨基己酸作为共晶配体,与乳清酸进行共结晶研究,成功获得5 种新的共晶物。共晶物粉末体外溶解实验表明,共晶物与乳清酸自身相比,溶解性均得到明显提高。
尽管大量文献已经证明COSMO-RS方法在CCF的预测、筛选方面的独特优势,但COSMO-RS方法仅能预测分子间的亲和力,不能完全确保共晶的形成。事实上,除了共晶外,实验筛选后的最终结果可能是共无定形状态,或者仅仅是这2种化合物的物理混合物(没有2个分子之间的相互作用)[67]。因此,虽然理论计算可以为快速筛选提供方向和选择,但仍需实际的实验操作来验证。
4 结束语
近年来,随着学科之间不断地交叉、渗透,量子化学理论计算逐渐与实验化学相结合,成为新的研究热点。基于量子化学的药物共晶虚拟筛选策略可以极大地减少实验筛选次数、缩小筛选范围、加快实验进程、提高药物共晶的筛选成功率。值得注意的是,任何基于理论计算初期的预测与实验实际筛选结果都存在一定的偏差,需要大量的实验结果补充数据库,对计算理论不断优化,才能实现高准确率的快速高通量筛选。不断优化、创新理论计算方法仍是实现高效共晶筛选面临的巨大挑战。
