三维荧光光谱结合二阶校正算法对两种植物病原菌快速定性定量分析
2022-04-26蔡鲁宁刘雪茹古绍彬
蔡鲁宁,刘雪茹,陈 磊,李 欣,2,3, ,古绍彬,
(1.河南科技大学食品与生物工程学院,河南 洛阳 471023;2.河南科技大学微生物资源开发与利用校级重点实验室,河南 洛阳 471023;3.河南省食品微生物工程技术研究中心,河南 洛阳 471023)
传统的微生物鉴定方法主要是通过形态学观察并辅以生化实验对微生物种属进行判断[1],存在操作复杂繁琐,耗时费力,准确率不高等缺点。分子生物学鉴定方法虽然能够实现准确分类,但需要专业的机构和人员且操作成本高。使用荧光检测仪器快速获得微生物样本的数据信息,并依据微生物独特的信息表达实现微生物的快速分类和定量,是一种新颖的研究角度。三维荧光光谱技术具有选择性好、灵敏度高、测试快速等优点。二阶校正可以在干扰存在的情况下对组分进行定性定量分析,这一特点也被称为二阶优势[2]。三维荧光光谱结合二阶校正算法目前已在农药检测[3]、食用植物油真伪鉴别[4]、酚类物质检测[5]、原油检测[6−7]、水环境[8−9]、医药[10]等众多领域中广泛应用。
荧光光谱分析技术在微生物鉴别方面已有研究,例如,通过紫外线激发的固有荧光可实现细菌和酵母物种的区分[11],KANG等[12]使用三维荧光光谱实现口腔中7种不同细菌的鉴定。此外,内源荧光法作为一种更便捷、灵敏的研究方法,在细菌鉴别方面受到广泛关注[13]。目前,利用三维荧光光谱技术对单一体系下的纯种微生物进行检测鉴定的研究较多。然而,在对含有多种微生物的复杂体系中目标微生物的定性和定量分析鲜有研究。黄瓜细菌性角斑病与霜霉病症状表现类似,给黄瓜细菌性角斑病的诊断和预防增加了难度,黄瓜细菌性角斑病与霜霉病的区分是诸多学者研究黄瓜病害的热门话题[14],黄瓜细菌性角斑病的病原菌是丁香假单胞菌黄瓜致病变种(Pseudomonas syringaepv. Lachrymans,PSL),它的检测通常依据形态学、生理生化检验和分子生物学鉴定等方法实现[15],三维荧光光谱分析技术能否成为PSL的检测方法仍值得探究;小麦赤霉病是由多种镰孢菌侵染所引起的小麦病害,禾谷镰孢菌(Fusarium graminearum)是其中之一[16],赤霉病能够使小麦种子发芽生长受阻[17],病麦中含有多种真菌毒素包括脱氧雪腐镰刀菌烯醇(DON)和玉米赤霉烯酮(ZEN)等[18−19],能导致人畜中毒,因此,探寻快捷而又经济的病原菌检测方法对于小麦赤霉病的预防具有重大意义。
本文以黄瓜细菌性角斑病病原菌丁香假单胞菌黄瓜致病变种(Pseudomonas syringaepv.Lachrymans-8,PSL-8)[20−21]和小麦赤霉病病原菌禾谷镰孢菌(Fusarium graminearum-ACCC37687)[22−23]为研究对象,运用三维荧光光谱分析技术快速鉴别植物真菌病害和细菌病害的病原微生物,探索三维荧光光谱分析技术快速识别植物真菌细菌病害的可行性。通过收集梯度混合菌液样本的三维荧光光谱数据,探究二阶校正算法对三维荧光光谱数据进行解析、提取特征激发和特征发射波长的能力,对特征波长荧光强度数据和菌液在600 nm波长下的吸光度(OD600)进行多元线性回归从而建立浓度预测模型,使用留一法交叉验证(Leave-One-Out Cross Validation,LOOCV)衡量模型预测性能,进而实现复杂的菌液混合体系下对单组分的定性定量分析。
1 材料与方法
1.1 材料与仪器
LB液体培养基:酵母浸粉5 g、胰蛋白胨10 g、氯化钠10 g 北京奥博星生物技术有限责任公司;PDA斜面培养基 青岛海博生物技术有限公司;丁香假单胞菌PSL-8 中科院植保所赵廷昌教授惠赠;禾谷镰孢菌 中国农业微生物菌种保藏管理中心,菌株编号:ACCC37687。
Smart Fluo-Pro Flex One型三维荧光光谱仪北京卓立汉光仪器有限公司; LDZX-30KBS型立式压力蒸汽灭菌锅 上海申安医疗器械厂;SW-CJ-1FD型洁净工作台 苏州安泰空气技术有限公司;H1650-W型医用离心机 湖南湘仪实验室仪器有限公司;THZ-92C台式恒温振荡器 上海跃进医疗器械有限公司;DNP-9022型电热恒温培养箱 北京市永光明医疗仪器厂;722s可见分光光度计 上海精密科学仪器有限公司。
1.2 实验方法
1.2.1 单菌液制备
1.2.1.1 丁香假单胞菌菌液制备 将活化后的丁香假单胞菌菌液按百分之一接种量接种到LB液体培养基,28 ℃、220 r/min振荡培养15 h,菌液离心7000 r/min,10 min,弃上清,用无菌双蒸水制成菌悬液,检测吸光度OD600,用无菌双蒸水将OD600调整到0.5[24]。
1.2.1.2 禾谷镰孢菌孢子悬液制备 在超净工作台中挑取小块禾谷镰孢菌菌块接种到灭菌的PDA斜面培养基上,28 ℃培养3~5 d,将培养好的禾谷镰孢菌菌种斜面,倒入5 mL无菌水,轻轻将PDA培养基表面的孢子刮下,将孢子悬液置于已灭菌的50 mL三角瓶内,瓶内预先放置数粒无菌玻璃球,采用充分振荡后灭菌的纱布(180 r/min,10 min,28 ℃,八层纱布)进行过滤,即得孢子悬浮液。一组实验需要9~10个禾谷镰孢菌菌种斜面。检测孢子悬浮液吸光度OD600,用无菌双蒸水将OD600调整到0.5[25]。
1.2.2 混合菌液制备 将制备得到的丁香假单胞菌菌液(OD600=0.5)和禾谷镰孢菌孢子悬液(OD600=0.5)梯度混合,每个样本期望OD600如表1所示共11个梯度。后续建立浓度预测模型需要扩大样本量,为了扩大样本量同时验证低浓度下混合菌液荧光的规律性,建模时混合菌液OD600从0.5调整到0.2,括号内为建模时每个样本的期望OD600,收集40组混合菌液样本三维荧光光谱信息用于建模。设置激发波长范围为200~450 nm,步长为5 nm,共51个激发波长;发射波长范围为260~800 nm,步长0.4 nm,共1351个激发波长,积分时间是1 s。按照以上设置扫描三维荧光光谱,每一组光谱数据量为11×51×1351,将光谱数据输入MATLAB软件进行预处理、数据解析和建模。

表1 混合菌液浓度梯度设置及期望OD600 Table 1 Concentration gradient setting and expected OD600 of mixed bacteria solution
1.3 数据处理
1.3.1 荧光数据预处理 对采集到的荧光光谱数据进行预处理来消除瑞丽散射的干扰。瑞丽散射一般在发射波长等于1倍或2倍激发波长区域及其临近区域(±10~15 nm左右)[7]。先将瑞丽散射区域数据扣除,采用Delaunay三次多项式插值方法,以瑞丽散射相邻区域的数据为基准进行三次多项式插值。然后使用(Savizkg-Golag,SG)多项式曲面平滑[26]来消除图谱中的噪音毛刺,并将预处理后的三维荧光光谱数据用于后续分析。
1.3.2 核心一致性诊断(CORCONDIA) 正确的组分数对于平行因子分析(Parallel Factor Analysis,PARAFAC)算法提取正确的特征波长起到关键的作用,CORCONDIA法[27]通过计算Tucker3模型中的超对角矩阵与最小二乘拟合阵之间的相近程度来估算组分数,相近程度用核心一致值来衡量。核心一致函数计算公式如下:
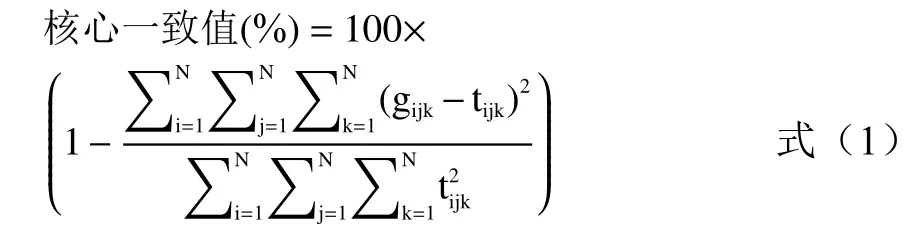
式中,gijk是Tucker3模型超对角矩阵元素;tijk为单位超对角矩阵;N是模型组分数;i、j、k分别表示三维荧光数据阵对应的第i个激发波长、第j个发射波长和第k个混合菌液样本。
当预测组分数N小于等于正确组分数时,计算得到的核心一致值越接近100%,此时模型符合三线性;N大于正确组分数时,计算得到的核心一致值接近0,模型偏离三线性。当组分数取到N时核心一致值大于60%,此时N-1就是正确组分数[2]。
1.3.3 三线性模型 将菌种荧光实验设定为I个激发波长和J个发射波长,对梯度浓度下的K个样本进行三维荧光光谱数据收集,得到大小为I×J×K的三维数据阵X[3]。三维数据阵的元素(i,j,k)设置为Xijk,则Xijk的表达式如下:

式中,ain是相对激发矩阵A(I×N)中的元素,bjn是相对发射矩阵B(J×N)中的元素,ckn是相对浓度阵C(K×N)中的元素。eijk是相对残差数阵E(I×J×K)中的元素。N是对样品体系中贡献响应信号的所有组分数,包括感兴趣组分、仪器噪声干扰组分和样品溶液杂质干扰组分[28]。
1.3.4 平行因子分析(PARAFAC) Harshman在1970年提出了平行因子分析(Parallel Factor Analysis,PARAFAC)算法[29],通过对以下目标函数的最小化求解相对激发矩阵A、相对发射矩阵B、相对浓度阵C:

PARAFAC算法同时进行的另外一个过程是将三维数据阵展开成二维数据阵,分别沿三个方向进行交替分解[30]:
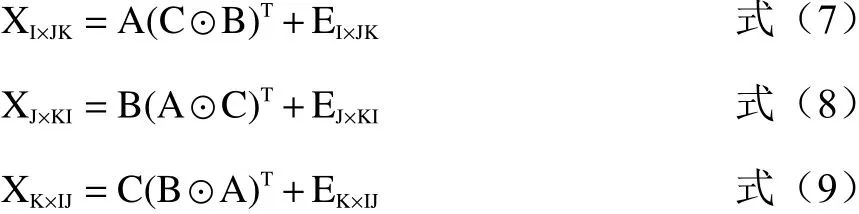
以上公式最小化可得:
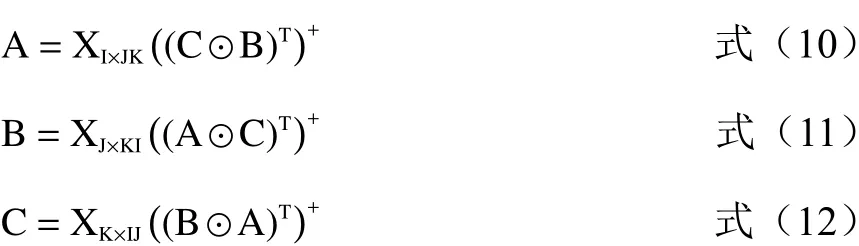
迭代计算过程如下:
第一步:确定样本体系的组分数N;第二步:相对激发矩阵A和相对发射矩阵B的初始化;第三步:根据相对激发矩阵A和相对发射矩阵B计算相对浓度阵C;第四步:根据对相对发射矩阵B和相对浓度阵C计算相对激发矩阵A,并且将相对激发矩阵A归一化;第五步:根据相对激发矩阵A和相对浓度阵C计算相对发射矩阵B,并且将相对发射矩阵B归一化;第六步:根据相对激发矩阵A和相对发射矩阵B计算相对浓度阵C,重复第4~6步,直到(SSRE(m)−SSRE(m−1))/SSRE(m−1)≤1×10−6
1.3.5 交替三线性分解(ATLD) 吴海龙在1998年提出了交替三线性分解(Alternating Trilinear Decomposition,ATLD)算法[31],相比于PARAFAC算法,ATLD提出了切片矩阵和广义逆运算的思想,将三维数据阵沿三个方向的切片矩阵为单位对相对激发矩阵A、相对发射矩阵B、相对浓度阵C进行分解解析,如下:

1.3.6 自加权交替三线性分解(SWATLD) 自加权交替三线性分解(Self-weighted Alternating Trilinear Decomposition,SWATLD)是在ATLD基础上进行了改进,将每个矩阵的迭代更新公式优化[7],例如对相对激发矩阵A的更新如下:

SWATLD与ATLD思路一致,因此具有ATLD具有的对组分不敏感,收敛快的优点,同时缓解了ATLD对背景噪声信号敏感的缺点。
1.3.7 交替惩罚三线性分解(APTLD) 交替惩罚三线性分解(Alternating Penalty Trilinear Decomposition,APTLD)是将平行因子分析(PARAFAC)和自加权三线性分解(SWATLD)两种算法进行组合优化[4],在三线性模型目标函数中引入约束条件,将求解问题转变为约束条件下的最优解问题,同时引入惩罚项将有约束问题变成无约束问题[32]。以相对激发矩阵A为例:
矩阵A元素残差平方和定义为:

式中:F为三维数据阵X中的主因子数;A是相对激发矩阵、B是相对发射矩阵、C是相对浓度阵;Xi..、为三维数据阵X的第i个水平矩阵(J×K);ai为载荷阵A的第i行矩阵;diag(ai)、为ai对应的U×U阶对角矩阵;U是所有对荧光做出贡献的组分数。
相对激发矩阵A定义为:
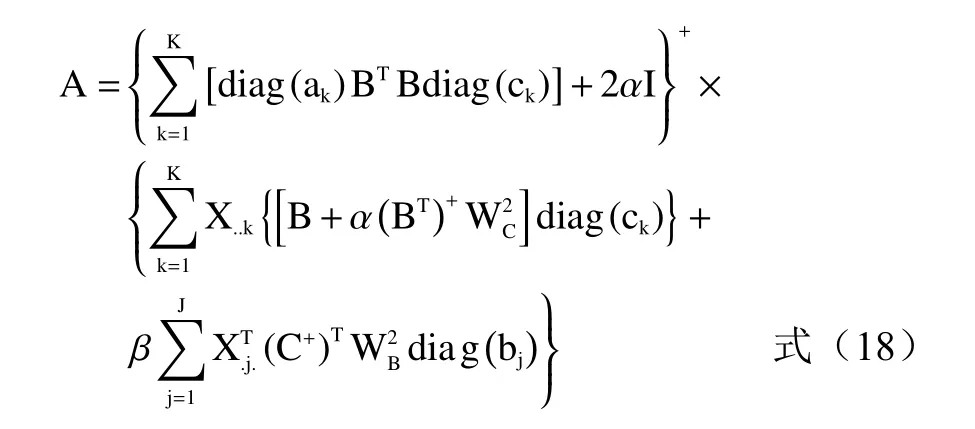
式中:A+、B+、C+分别为A、B、C的Moore-Penose广义逆矩阵,上标+为广义逆;WA、WB、WC为权重矩阵;α、β为平行因子分析(PARAFAC)与自加权三线性分解(SWATLD)的权重。
1.3.8 偏最小二乘回归系数法(PLS) 偏最小二乘回归系数法(Partial Least Squares,PLS)分别对混合菌液样本特征激发和特征发射波长进行提取[33−34]。偏最小二乘回归系数法是通过分别构建禾谷镰孢菌和丁香假单胞菌混合菌液下梯度上升和梯度下降的浓度值对激发光谱和发射光谱的回归模型,波长的回归系数代表该波长的贡献比重,回归系数的绝对值越大,代表该波长对回归模型预测性能的影响程度越大。
1.3.9 留一法交叉验证(LOOCV) 留一法交叉验证(Leave-One-Out Cross Validation,LOOCV)可以验证混合菌液浓度预测模型的性能,每次只留下一个样本做测试集,其它样本做训练集,直至所有样本均充当过测试集和验证集,模型预测能力由相关系数R、交叉验证决定系数R2cv、预测均方根误差RMSEP来衡量[35]。相关系数R和交叉验证决定系数R2cv越接近1、预测均方根误差RMSEP越小,表明浓度预测模型的性能越好,准确率越高,模型得到的预测值越接近真实值。
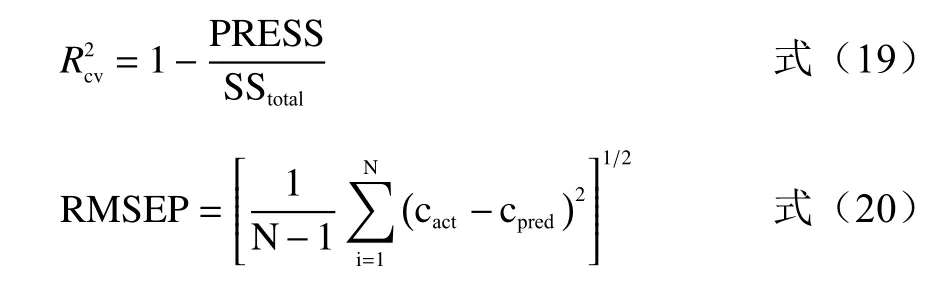
式中PRESS是预测残差平方和,SStotal是总平方和,cact和cpred分别是实际样本浓度和预测样本浓度。
2 结果与分析
2.1 荧光数据预处理结果
图1(A)是OD600=0.5的丁香假单胞菌和禾谷镰孢菌混合菌液的原始三维荧光光谱图,图1(B)是去除瑞丽散射再经过SG平滑处理后的三维荧光光谱图。通过前后对比发现,采集混合菌液的三维荧光数据时出现的瑞丽散射强度非常高以至于产生瑞丽墙,位于等高线图的右下方,原始三维荧光光谱的特征信息受到瑞丽散射形成的背景墙的干扰而难以凸显真实的三维荧光光谱信息。由图1(B)可以发现,去除瑞丽散射再经过SG平滑处理后,右下方的瑞丽散射墙被消除,混合菌液的特征峰位置信息得以呈现,完整保留荧光信息的同时能够有效消除瑞丽散射带来的不良影响,为后续特征信息的提取工作打下良好的基础。

图1 混合菌液SG平滑处理前和SG平滑处理后的光谱等高线图Fig.1 Spectral contour map of mixed bacterial solution before and after SG smoothing treatment
2.2 单一菌种的三维荧光光谱图
图2(A)是OD600=0.5的丁香假单胞菌经过SG平滑处理后的光谱图,图2(B)是OD600=0.5的禾谷镰孢菌经过SG平滑处理后的光谱图。丁香假单胞菌和禾谷镰孢菌的波峰位置和荧光强度清晰可见,差异明显,说明丁香假单胞菌和禾谷镰孢菌在混合菌液定性分析时相互干扰较小。丁香假单胞菌波峰位置为激发/发射(Ex/Em)=285~290 nm/336~360 nm,禾谷镰孢菌波峰位置为激发/发射(Ex/Em)=340~390 nm/410~470 nm。为实现混合菌种定量分析,需确定单一菌种所对应出峰位置,并与各类方法分解光谱图数据得到的每一组分出峰位置做出比较,依此判断每一组分所对应的菌种类别。

图2 丁香假单胞菌(PSL-8)和禾谷镰孢菌(ACCC37687)使用SG平滑处理后的光谱等高线图Fig.2 Spectral contour map of Pseudomonas syringae pv.Lachrymans-8 and Fusarium graminearum- ACCC37687 smoothed by SG
2.3 组分数确定
在进行三线性模型分解解析时,组分数的确定是分解过程中极为关键的一环,尤其是在使用平行因子分析时,需要找到合理的组分数[36]。虽然交替三线性分解对组分数不敏感,但是过高或者过低的组分数都会导致计算模型的误差[5],合适的组分数会使结果更加准确。故用核心一致性诊断(CORCONDIA)对预处理后的三维荧光光谱进行组分数的确定。图3反映了OD600=0.5混合菌液不同组分数所对应的核心一致值。
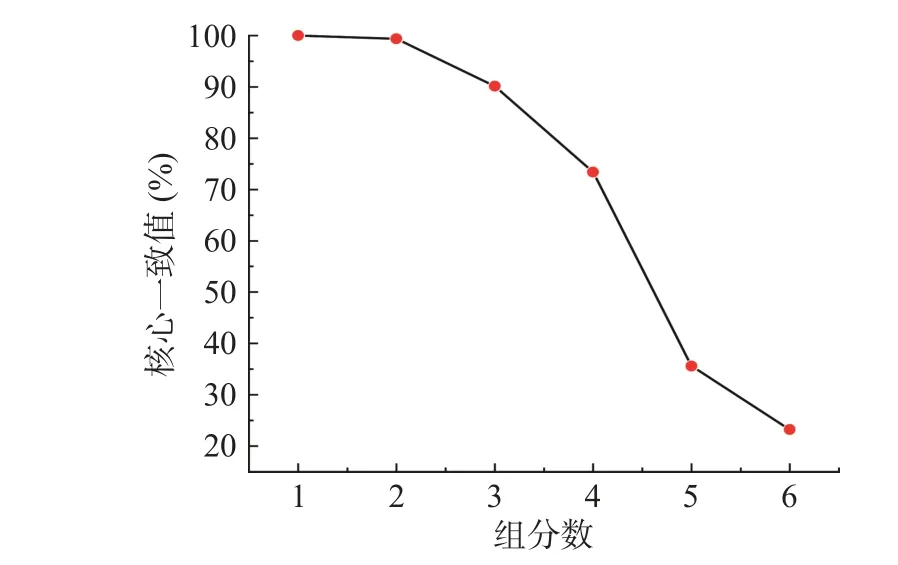
图3 不同组分数对应的核心一致值Fig.3 Core consistent values of different component scores
随着组分数的增加,核心一致值逐渐降低,1~4组分时核心一致值大于60%,当组分数大于4的时候核心一致值小于60%,故混合菌液的最佳组分数应是4。
2.4 使用二阶校正算法对混合菌液样品进行分析
将最佳组分数设定为4,分别使用交替三线性分解(ATLD)、平行因子分析(PARAFAC)、自加权三线性分解(SWATLD)、交替惩罚三线性分解(APTLD)对OD600=0.5混合菌液样品三维荧光光谱进行分解解析。会得到每一组分的相对激发强度光谱、相对发射强度光谱和组分相对浓度。四种组分的激发波长出峰位置清晰可见并且相互独立,未出现出峰位置交叠的情况,组分之间无相互干扰。分析激发发射光谱数据可以得到各个组分所对应的荧光峰出峰位置,汇总结果至表2。
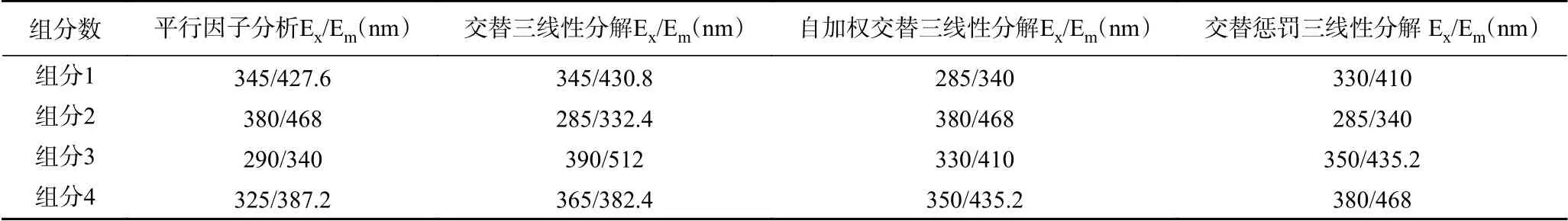
表2 在4组分下用二阶校正算法分解出的每一组分出峰位置Table 2 Peak position of each component decomposed by second-order correction algorithm under 4 components
将二阶校正算法分解光谱得到的各个组分的激发光谱发射光谱波峰位置与单一丁香假单胞菌和禾谷镰孢菌激发光谱发射光谱波峰位置相比较,对比每一组分波峰位置下的荧光强度变化趋势,结合菌种浓度变化规律,把既满足组分波峰位置和菌种波峰位置一致,又满足波峰位置荧光强度变化规律和菌种浓度变化规律一致的组分视为该菌种的特征组分,特征组分的波峰位置视为该菌种的特征波长。可以看出每一算法都能分解出一个符合丁香假单胞菌波峰位置和一个符合禾谷镰孢菌波峰位置的组分。由于采集三维荧光光谱时激发波长Ex的步长设置为5 nm,可以看出每种方法提取出的丁香假单胞菌的特征组分波峰位置只相差1~2个步长,波峰位置具有高度相似性,提取出的禾谷镰孢菌特征组分波峰位置也有相同的规律。将四种二阶校正算法提取出的丁香假单胞菌和禾谷镰孢菌相对激发光谱图(A)、相对发射光谱图(B)和组分相对浓度图(C)汇总进行对比,如图4。
由图4(A)和图4(B)可以看出四种二阶方法所提取出来的丁香假单胞菌的相对激发光谱峰形相似,相对发射光谱也具有高度一致性,出峰位置激发/发射(Ex/Em)=285(290)/332.4(340) nm。禾谷镰孢菌激发光谱和发射光谱峰形亦是如此,具有高度一致性,出峰位置激发/发射(Ex/Em)=380(390)/468(512) nm。由图4(C)可以看出,四种二阶方法预测浓度曲线趋势大致相同,可以分解出上升趋势和下降趋势两种曲线,符合实验设计的丁香假单胞菌浓度逐渐升高、禾谷镰孢浓度逐渐降低的规律。使用交替三线性分解(ATLD)、自加权交替三线性分解(SWATLD)、交替惩罚三线性分解(APTLD)获得的浓度曲线规律更明显,而平行因子分析(PARAFAC)在相同的荧光数据下的获得的浓度曲线在2号样品和8号样品出现异常浮动。

图4 二阶校正算法获得的OD600=0.5混合菌液丁香假单胞菌和禾谷镰孢菌相对激发强度光谱图(A)、相对发射强度光谱图(B)和组分相对浓度图(C)(N=4)Fig.4 Relative excitation intensity spectrum (A), relative emission intensity spectrum (B) and component relative concentration diagram (C) (N=4) of Pseudomonas syringae pv.Lachrymans-8 and Fusarium graminearum-ACCC37687 in mixed bacterial solution with OD600=0.5 obtained by the secondorder correction algorithm method
2.5 使用偏最小二乘回归系数法(PLS)对混合菌液样品进行分析
用偏最小二乘回归系数法(PLS)提取OD600=0.5混合菌液的特征激发波长和特征发射波长,得到混合菌液的前三组分权重系数图。分别按浓度逐渐上升和浓度逐渐下降对混合菌液三维荧光光谱数据使用PLS方法,结果如图5所示。
每个波长所对应回归系数绝对值的大小代表该波长对预测模型性能影响的程度[37]。由图5可知,第1组分和第2组分不同波长所对应的回归系数波动范围较小,不能较好的体现不同样本之间的差异。因此选取波动幅度更大、能更好的体现样本差异的第3组分作为特征组分,该组分回归系数的波峰波谷对应的波长为特征波长。由图5(A)可得,按浓度上升使用PLS得到的特征激发波长分别为270、280、295、315、320、345、415 nm。由图5(B)可得特征发射波长分别为361.6、416、515.6 nm。由图5(C)可得,按浓度下降使用PLS得到的特征激发波长分别为285、295、315、320、340、415 nm。由图5(D)可得特征发射波长为360.8、415.2、511.2 nm。
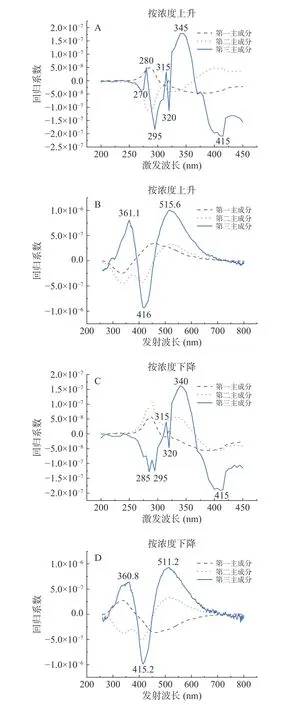
图5 激发波长权重系数图和发射波长权重系数图Fig.5 Excitation wavelength weight coefficient diagram and emission wavelength weight coefficient diagram
先将筛选出来的特征波长与单一丁香假单胞菌和禾谷镰孢菌波峰位置进行比对,再通过对比各特征波长下荧光强度变化趋势,结合菌种浓度变化规律,把既满足特征波长位置和菌种波峰位置一致,又满足该波长下荧光强度变化规律和菌种浓度变化规律一致的波长叫做该菌种的特征波长,筛选得出激发/发射(Ex/Em)=280 nm/361.6 nm和激发/发射(Ex/Em)=295 nm/361.6 nm是丁香假单胞菌的特征激发发射波长位置。激发/发射(Ex/Em)=340 nm/511.2 nm和激发/发射(Ex/Em)=415 nm/511.2 nm是禾谷镰孢菌的特征激发发射波长位置。PLS提取得到的特征波长用于后续混合菌液的定量分析。
2.6 光谱拟合定性分析
为了验证上述判断,并且考察各个算法定性分析能力,将每个算法分解得到的特征激发光谱和特征发射光谱与单一丁香假单胞菌和单一禾谷镰孢菌的真实光谱,归一化后进行拟合。
图6、图7分别是各个算法分解混合菌液三维荧光数据阵得到的激发光谱和发射光谱与单一丁香假单胞菌和禾谷镰孢菌的真实激发发射光谱两者拟合得到的。可以看出,平行因子分析(PARAFAC)、交替三线性分解(ATLD)、自加权交替三线性分解(SWATLD)、交替惩罚三线性分解(APTLD)对丁香假单胞菌都具有良好的定性效果,不论激发光谱还是发射光谱都可以分解出一个和丁香假单胞菌光谱形状高度一致的组分,验证了前面的推论。
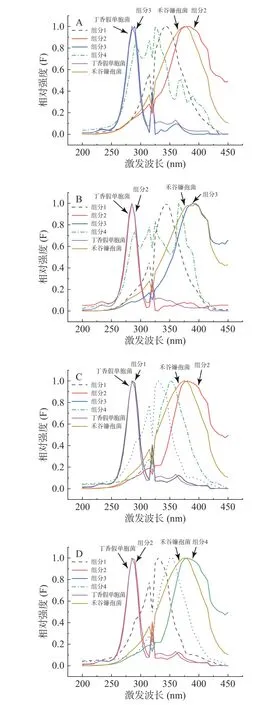
图6 平行因子分析(A)、交替三线性分解(B)、自加权交替三线性分解(C)、交替惩罚三线性分解(D)方法获得的激发光谱和丁香假单胞菌(PSL-8)、禾谷镰孢菌(ACCC37687)的真实激发光谱Fig.6 The excitation spectra obtained by PARAFAC (A),ATLD (B), SWATLD (C), APTLD (D) and the real excitation spectra of Pseudomonas syringae pv. Lachrymans-8 and Fusarium graminearum-ACCC37687
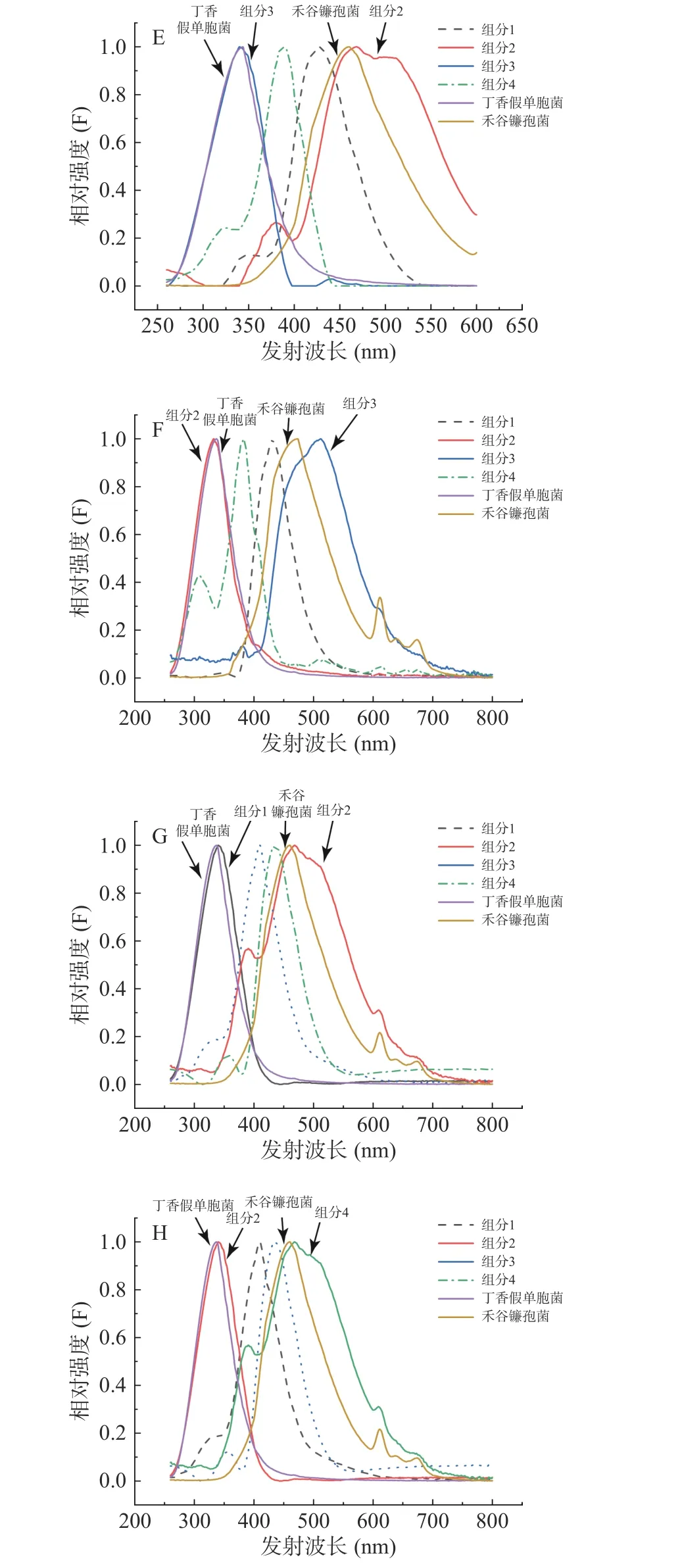
图7 平行因子分析(E)、交替三线性分解(F)、自加权交替三线性分解(G)、交替惩罚三线性分解(H)方法获得的发射光谱和丁香假单胞菌(PSL-8)、禾谷镰孢菌(ACCC37687)的真实发射光谱Fig.7 The emission spectra obtained by PARAFAC (E), ATLD(F), SWATLD (G), APTLD (H) and the real emission spectra of Pseudomonas syringae pv. Lachrymans-8 and Fusarium graminearum- ACCC37687
平行因子分析(PARAFAC)、交替三线性分解(ATLD)、自加权交替三线性分解(SWATLD)、交替惩罚三线性分解(APTLD)对禾谷镰孢菌的鉴定效果较差,无峰形完全一致的组分与之对应。自加权交替三线性分解(SWATLD)、交替惩罚三线性分解(APTLD)有部分与禾谷镰孢菌真实光谱高度重叠的波形,因此效果优于平行因子分析(PARAFAC)、交替三线性分解(ATLD)。与禾谷镰孢菌真实光谱相比,四种二阶校正算法分解出来的禾谷镰孢菌对应组分的光谱峰形均发生后移。将单一禾谷镰孢菌特征激发波长和特征发射波长处的光谱与混合菌液下禾谷镰孢相同波长处的光谱进行拟合,发现菌液混合后禾谷镰孢菌峰形确实发生变化。有文献表明峰形后移的原因可能有多种,色氨酸与配体结合,或蛋白质与蛋白质结合以后发射波长会发生位移[13],峰形后移的原因需要进一步探索验证。同时每种算法都会有两种未知干扰组分,推测是两种菌共同拥有的荧光基团或者是出峰位置相近的荧光基团。
2.7 留一法交叉验证(LOOCV)多元线性回归构建浓度预测模型
浓度预测模型的构建需要大批量实验作为测试集和验证集,故将混合菌液OD600变更为0.2,同时也能验证高浓度样本提取出来的特征波长在样品浓度降低时是否依旧有规律。总共做了40组混合菌液样本,每组11个浓度梯度样本,总共440个样本三维光谱数据。应用交替惩罚三线性分解(APTLD)、自加权交替三线性分解(SWATLD)、平行因子分析(PARAFAC)、交替三线性分解(ATLD)、偏最小二乘回归系数法(PLS)提取出来的丁香假单胞菌主荧光峰位置的激发波长和发射波长分别是激发/发射(Ex/Em)=285 nm/340 nm、290 nm/340 nm、285 nm/332.4 nm、280 nm/361.6 nm、295 nm/361.6 nm。提取出来的禾谷镰孢菌主荧光峰位置激发波长和发射波长分别是激发/发射(Ex/Em)=380 nm/468 nm、
390 nm/512 nm、340 nm/511.2 nm、415 nm/511.2 nm。使用多元线性回归函数对样本OD600和筛选得到的菌种对应特征波长处的荧光强度构建回归模型,将混合菌液每种菌种的期望OD600值作为因变量Y,将各类方法挑选出的特征波长的荧光强度作为自变量X,分别得到丁香假单胞菌和禾谷镰孢菌两种菌的浓度预测模型, 再使用留一法交叉验证(LOOCV)浓度预测模型的准确性,分别计算单一方法和综合方法下模型的相关系数R、交叉验证决定系数R2cv、预测均方根误差RMSEP,依此选出最佳的模型方案,结果汇总见表3,最佳预测模型下,两种菌浓度预测结果见表4。
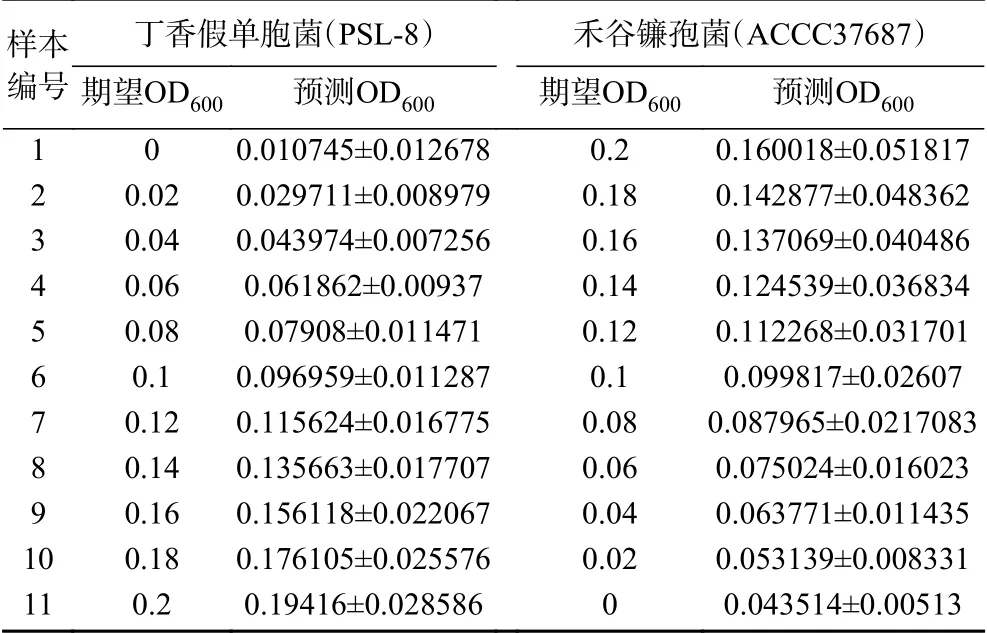
表4 两种菌的浓度预测结果Table 4 Predicted results of two bacterial concentrations
由表3可知,使用单一方法建立浓度预测模型时,交替三线性分解(ATLD)建立的丁香假单胞菌浓度预测模型效果最好,自加权交替三线性分解(SWATLD)、交替惩罚三线性分解(APTLD)的浓度预测效果次之,平行因子分析(PARAFAC)和偏最小二乘回归系数法(PLS)效果最差。偏最小二乘回归系数法(PLS)建立的禾谷镰孢菌浓度预测模型效果最好,交替三线性分解(ATLD)的浓度预测效果次之,平行因子分析(PARAFAC)、自加权交替三线性分解(SWATLD)、交替惩罚三线性分解(APTLD)的效果最差。将所有方法提取的特征波长综合应用从而建立的丁香假单胞菌和禾谷镰孢菌浓度预测模型效果最好。丁香假单胞菌和禾谷镰孢浓度预测模型如下:
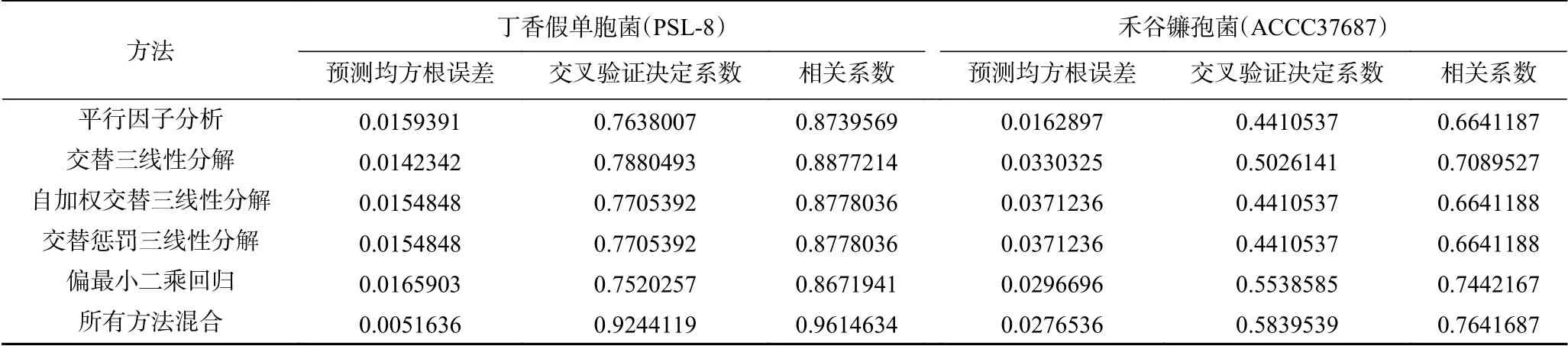
表3 模型稳定性参数汇总Table 3 Summary of model stability parameters
混合菌液丁香假单胞菌浓度预测模型:

式中:YPSL-8是丁香假单胞菌的OD600预测值;X1是ATLD筛选出来的组分2特征波长处的荧光强度;X2是PARAFAC筛选出来的组分3特征波长处的荧光强度;X3和X4是PLS按菌液浓度线性上升筛选出来的两个特征波长处的荧光强度。APTLD、SWATLD筛选得到的丁香假单胞菌特征波长相同,X5是APTLD(SWATLD)筛选出来的特征波长处的荧光强度。
混合菌液禾谷镰孢菌浓度预测模型:

式中:Y禾谷镰孢是禾谷镰孢菌的OD600预测值;X6是ATLD筛选出来的组分3特征波长处的荧光强度;X7是PARAFAC筛选出来的组分2特征波长处的荧光强度;APTLD、SWATLD筛选得到的禾谷镰孢菌特征波长与PARAFAC筛选出来的特征波长相同;X8和X9是PLS按菌液浓度线性下降筛选出来的两个特征波长处的荧光强度。
使用留一法交叉验证(LOOCV)浓度预测模型的准确性,得到混合菌液下丁香假单胞菌浓度预测模型的交叉验证决定系数R2cv=0.92441191,预测均方根误差RMSEP=0.005163633,相关系数R=0.961463421。混合菌液下禾谷镰孢菌浓度预测模型的交叉验证决定系数R2cv=0.583953931,预测均方根误差RMSEP=0.027653679,相关系数R=0.764168784。两者比较可以看出丁香假单胞菌浓度预测模型的预测效果明显好于禾谷镰孢菌浓度预测模型。推测禾谷镰孢菌浓度预测模型预测效果不佳的原因可能在于真菌本身不适用于比浊法来衡量真菌孢子浓度,当使用OD600作为真菌孢子浓度衡量标准时,实验操作本身带来的操作误差较大,不同样本相同OD600下的真菌孢子悬液的孢子浓度可能有所不同,这样极大的影响了荧光的规律性。
3 结论
本文采集丁香假单胞菌和禾谷镰孢菌梯度混合菌液的三维荧光光谱数据,结合二阶校正算法及一阶校正算法筛选出菌种特征波长, 通过对特征波长荧光强度数据和菌液在600 nn波长下的吸光度(OD600)进行多元线性回归从而建立浓度预测模型。实验表明,通过多元线性归建回立的丁香假单胞菌浓度预测模 型(R2cv=0.92441191,RMSEP=0.005163633,R=0.961463421)比禾谷镰孢菌浓度预测模型(R2cv=0.583953931,RMSEP=0.027653679,R=0.764168784)效果更佳,定性定量分析结果符合预期。而利用荧光光谱技术对真菌的定量分析可能存在一定的局限性,禾谷镰孢菌浓度预测模型对禾谷镰孢菌的浓度预测误差明显,梯度混合菌类别太少,这进一步限制了该方法的普适性和推广。所得实验结果对该研究的价值与预期有所差别,但实验方法对研究利用荧光光谱技术对真菌的定量分析提供了利用价值。
