超声波处理对兔毛角蛋白组成与结构的影响
2022-04-24王晓清史志铭李晓宇
王晓清, 史志铭, 李晓宇
(1. 内蒙古工业大学 轻工与纺织学院, 内蒙古 呼和浩特 010080; 2. 内蒙古工业大学 材料科学与工程学院, 内蒙古 呼和浩特 010051)
兔毛作为典型的特种动物纤维,资源丰富,价格低廉,是一种由角蛋白、微量色素以及灰分等组成的蛋白质纤维,其中角蛋白含量高达90%以上[1]。角蛋白由多种氨基酸通过二硫键、氢键、离子键和疏水作用连接形成四级结构[2],使兔毛具有稳定的化学性能和较强的力学性能[3]。然而由于特殊的髓质层和鳞片层结构,导致兔毛可纺性能较差,每年在纺纱过程中都会产生大量兔毛废料,造成严重的资源浪费和环境污染,因此,将兔毛废料转化为有用的角蛋白材料引起越来越多的关注。
角蛋白具有生物相容性和生物可降解性,此外提取的角蛋白能够形成调节细胞识别和行为的自组装结构,因此,被认为是一种潜在的生物材料新资源,可用于组织医学领域[4-5]。鉴于以上优点,且动物毛发资源丰富,近年来许多研究采用化学水解、氧化、还原等方法提取角蛋白[6-8],还有采用蒸汽闪爆辅助碱液以及离子液体提取角蛋白的报道[9-10]。角蛋白提取的原理主要是破坏纤维中胱氨酸的二硫键(S—S)形成半胱氨酸(—SH),使蛋白质大分子链断裂,进而破坏角蛋白的各级结构,最终使纤维解体。通常采用增加化学试剂用量或提高温度使二硫键的破坏程度增加,进而提高角蛋白的提取效率,但这会导致大分子主链上更多的肽键水解生成大量小分子多肽,在一定程度上限制了角蛋白的再次利用。
兔毛髓质层发达,结构均匀,分级排列,这种结构有利于超声波空化作用[11],因此,本文采用超声波辅助还原法对兔毛角蛋白进行提取,在一定程度上通过增加物理作用,降低化学作用对酰胺键的破环,在保证角蛋白提取效率的基础上制得分子质量高的角蛋白,对提取的角蛋白结构进行表征,研究超声波处理对兔毛角蛋白组成和结构的影响。
1 实验部分
1.1 实验材料和仪器
材料:兔毛,采自中国甘肃天水兔子养殖场,品种为德系安哥拉长毛兔;尿素,天津大茂化学试剂有限公司;亚硫酸氢钠,天津化学试剂有限公司;十二烷基硫酸钠(SDS),山东优索化工科技有限公司;石油醚、无水乙醇、氢氧化钠、氢氧化锂和盐酸,国药集团化学试剂有限公司;丙烯酰胺、N,N′-亚甲基双丙烯酰胺、过硫酸铵、甘油、无水甲醇、冰乙酸、溴酚蓝、甘氨酸(Gly),天津市福辰化学试剂有限公司;5,5′-二硫代-双-(2-硝基苯甲酸)(DTNB)、三羟甲基氨基甲烷(Tris),恒远博泰生物科技有限公司;考马斯亮蓝R-250,Sigma-Aldrich公司;四甲基乙二胺(TEMED)、 β-巯基乙醇、乙酸钠缓冲液和Tris-HCl缓冲液,上海麦克林生化科技有限公司;乙二胺四乙酸(EDTA),天津市化学试剂一厂;截留分子质量为8~14 ku的透析袋,北京索莱宝科技有限公司。
仪器:LGJ-12A冷冻干燥机,北京四环起航科技有限公司;L-8900氨基酸分析仪,日本日立公司;LC-20AT高效液相色谱仪、UV-2700紫外-可见分光光度计、IR Affinity-1傅里叶红外光谱仪,日本岛津公司;DYCZ-24DN电泳仪,北京六一生物科技有限公司;inVia Reflex拉曼光谱仪,英国雷尼绍公司;D/MAX-2500/PC X射线衍射仪,日本理学公司;G9800A荧光分光光度计,美国安捷伦科技公司;NanoBrook Omni激光粒度仪,美国布鲁克海文仪器公司。
1.2 兔毛角蛋白的提取
首先,将经石油醚和无水乙醇脱脂后的兔毛(RH), 采用超声波进行预处理一定时间,振荡频率为50 kHz,加热温度为60 ℃。然后,将预处理后的兔毛纤维(UT-RH)浸泡在尿素-亚硫酸氢钠-SDS混合溶剂体系中,调节溶剂pH值为9,加热至90 ℃反应4.5 h。之后,将溶解液经筛孔尺寸为 0.075 mm 的筛子过滤去除未溶解兔毛得到兔毛角蛋白溶液,抽滤后采用透析管在蒸馏水中透析48 h,以去除反应中形成的小分子物质和盐。最后,将透析后的角蛋白溶液在-80 ℃冷冻干燥获得兔毛角蛋白粉末(RHK)。 超声波处理时间分别为0、1、2、3、4 h, 制备的角蛋白试样依次记为UT-0、UT-1、UT-2、 UT-3、 UT-4。
1.3 测试及表征
氨基酸含量测试:采用氨基酸分析仪对兔毛和角蛋白的氨基酸含量进行测定。将样品置于6 mol/L 盐酸中,于110 ℃氮气气氛下水解24 h。水解氨基酸由羟基琥珀酰氨基甲酸酯衍生,然后反相柱洗脱。采用高效液相色谱仪在254 nm处检测洗脱液,用外标液(氨基酸标准溶液)通过峰面积计算样品测定液中氨基酸的含量。
色氨酸的测定:按照GB/T 15400—2018《饲料中色氨酸的测定》,将样品置于4 mol/L氢氧化锂溶液中,在110 ℃氮气气氛下水解20 h,用乙酸钠缓冲溶液(pH值为4.5)将水解液定量地转移至25 mL容量瓶中,并用上述缓冲溶液定容,经过0.45 μm滤膜后利用高效液相色谱仪测定。荧光检测器的激发波长为283 nm,检测发射波长为343 nm,采用外标法单点校正定量色氨酸的含量。
游离巯基含量测试:根据Iesel等[12]的方法进行游离巯基的测定。将15 mg角蛋白溶解于 5 mL 的Tris-Gly缓冲液(pH值为8.0)中,然后加入50 μL Ellman试剂(DTNB溶于Tris-Gly缓冲液,质量浓度为4 mg/mL),将配好的溶液置于25 ℃条件反应60 min,离心15 min,取上清液在412 nm处测定其吸光度,以含Ellman试剂的Tris-Gly 缓冲液为空白对照。巯基含量的计算公式为
式中:A为除去空白对照后样品在412 nm处的吸光度;D为稀释倍数;C为样品质量浓度,mg/mL。
角蛋白分子质量测定:利用电泳仪,采用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)测定兔毛角蛋白的分子质量分布[13]。RHK溶液与上样缓冲液(0.4 g SDS,20 mg溴酚蓝,2 mL甘油,1 mL 1 mol/L 的Tris-HCl(pH值为8),20 μL巯基乙醇混合定容至10 mL)以1∶1体积比混合均匀,煮沸5 min 使RHK变性。取20 μL变性RHK溶液加入预先配制好的Tris-HCl凝胶(分离胶含量为10%,浓缩胶含量为5%)中,在60 V电压下进行电泳,当样品进入分离胶后电压改为120 V。电泳分离后的凝胶用考马斯亮蓝R-250染色,最后用10%乙酸和5%甲醇洗脱,直到蛋白质区带清晰。低分子质量标准蛋白Marker在相同的电泳条件下运行。
二级结构测试:采用傅里叶红外光谱仪(FT-IR) 对兔毛及其角蛋白的二级结构进行表征,扫描范围为4 000~400 cm-1,扫描次数为40,分辨率为4.0 cm-1。采用Origin软件对样品进行基线校正、Savitsky-Golay函数平滑处理,再根据酰胺Ⅰ带吸收峰进行归一化处理,作其二阶导数和傅里叶去卷积曲线,用高斯函数解析酰胺I带,最后根据α-螺旋、β-折叠、β-转角、无规卷曲各二级结构积分面积计算得出其相对含量[14]。
(5) 逐步建立适用于轨道交通行业的复合材料标准体系,包括设计标准、材料评价标准、计算验证标准、生产工艺标准、质量检测标准和运用维护标准等,使复合材料在轨道交通领域的应用有据可依、规范化、系统化,从而促进轨道交通行业的节能减排和绿色转型升级。
拉曼光谱测试:采用氩离子激光器作为拉曼扫描光源进行测试,工作功率为10.2 mW,输出波长为633 nm,光谱范围为2 000~400 cm-1。
晶体结构测试:采用X射线衍射仪(XRD)测定兔毛及其角蛋白的晶体结构。测试条件:使用 Cu Kα 辐射源,管电压为40 kV,管电流为200 mA,扫描方式为连续扫描,扫描速度为3 (°)/min,扫描范围为5°~60°,步长为0.02°。利用MDI Jade软件对样品进行结晶度及晶面间距的计算。
三级结构测试:采用荧光分光光度计在室温下测定角蛋白的内源性荧光光谱,激发波长为 300 nm, 扫描范围为300~550 nm。
粒径与电位测试:采用激光粒度仪在25 ℃条件下,对兔毛角蛋白的粒径和电位进行测量。
2 结果与讨论
2.1 氨基酸分析
兔毛及其角蛋白的氨基酸组成如图1所示。可以看出,RH中谷氨酸(Glu)和胱氨酸(Cys)含量最多,精氨酸(Arg)、亮氨酸(Leu)、丝氨酸(Ser)和天冬氨酸(Asp)含量次之,而赖氨酸(Lys)、蛋氨酸(Met)、 组氨酸(His)和色氨酸(Trp)含量较低。与RH相比,UT-RH中大部分氨基酸组分没有变化,但含量均有所降低。Met含量降低最为明显,降低率高达18.50%,Cys和Lys含量降低也比较明显,降低率分别为6.58%和5.81%;只有His、Gly和苯丙氨酸(Phe)的含量出现小幅度增加。研究显示超声波处理蛋白质纤维,可使纤维的细胞间质和鳞片层发生不同程度的破坏,导致水溶性蛋白溶出,造成氨基酸含量减少[15]。超声波处理兔毛纤维的氨基酸测试结果显示,超声波处理不仅可使水溶性蛋白溶出,对稳定性较强的胱氨酸也有明显的破环作用。

图1 氨基酸含量分析结果Fig.1 Analysis result of amino acid content
与RH相比,兔毛角蛋白中胱氨酸变化最明显,兔毛中胱氨酸含量为11.74%,提取的兔毛角蛋白中胱氨酸含量最低为2.94%,约有70%的胱氨酸被破坏。同时,超声波处理时间不同,角蛋白胱氨酸含量也不相同,与UT-0试样相比,UT-1、UT-2试样的胱氨酸含量降低程度较小,当超声波处理3 h以上,胱氨酸含量损失增大。胱氨酸作为动物纤维中重要的一种氨基酸,通过二硫键的结合形成高级结构,维持蛋白质纤维的形态及力学性能。提取角蛋白的关键是打破胱氨酸的二硫键,在以前的研究中显示,超声波处理纤维在一定程度上可使纤维的结晶度降低,这样有利于还原剂很快进入纤维内部,与二硫键进行充分作用。由此可知,超声波处理兔毛纤维有利于角蛋白的提取。此外,角蛋白中Asp、Glu、丙氨酸(ALa)、Leu、酪氨酸(Tyr)、Arg都有明显的增加,反之Arg、Lys、脯氨酸(Pro)有所降低,且与超声波处理时间基本呈正相关,缬氨酸(Val)、Met、异亮氨酸(Ile)、His相对较为稳定,这与氨基酸的溶解度和热稳定性有关。最为特殊的是苏氨酸(Thr)的变化,与RH相比,超声波处理兔毛和角蛋白中Thr含量都有所下降,且变化基本相同,由此可知Thr几乎不受还原体系中化学试剂的影响。
不同种类氨基酸含量结果如表1所示。可以看出,与RH相比,超声波处理角蛋白中的总氨基酸含量降低,最高可达88.30%,这主要是由于碱性氨基酸和中性氨基酸减少,超声波处理时间增加,这2种氨基酸损失有所降低,相反兔毛角蛋白中酸性氨基酸则有少量增加。

表1 不同种类氨基酸含量Tab.1 Contents of different kinds of amino acids
2.2 游离巯基含量分析

图2 超声波处理时间对兔毛角蛋白胱氨酸 及游离巯基的影响Fig.2 Effect of ultrasonic treatment on Gys and free sulfhydryl groups of keratins
提取过程中,二硫键在亚硫酸氢钠的作用下断裂形成巯基,1 mol二硫键断裂生成2 mol半胱氨酸残基,但生成的巯基不稳定,极易被氧化形成新的二硫键生成胱氨酸,所以游离巯基和胱氨酸的变化情况有一定差别,游离巯基随超声波处理时间增加大幅增加,但在超声波处理的前2 h内,胱氨酸变化却不明显。巯基除再被氧化生成二硫键之外,有研究显示部分游离巯基在空气中被氧化生成砜类化合物[9],或形成挥发性硫化物,如硫化氢、挥发性有机化合物或风味物质,或者是生成硫胺酸等非常见的氨基酸,这也是游离巯基变化与胱氨酸变化不一致的原因。
2.3 分子质量及其分布分析
纤维状角蛋白是由沿单轴平行排列的多肽链段组成,经过超声波处理结合还原剂作用,二硫键断裂形成分子质量不同的低硫蛋白亚基和高硫蛋白亚基。在电泳过程中,由于分子质量大小不同,这些蛋白亚基就会在电泳图谱上分布在不同的位置[16]。采用SDS-PAGE分析了不同超声波处理时间提取的兔毛角蛋白的分子质量,结果如图3所示。

图3 兔毛角蛋白的 SDS-PAGE 图谱Fig.3 SDS-PAGE image of extracted rabbit hair keratin
电泳图谱显示,不同提取条件下RHK的分子质量分布不同。UT-0显示出较为分散的条带结构,角蛋白部分片段分子质量集中在14.4 ku以下,对应于蛋白质单体,这一系列低分子质量组分来自于嵌入中间纤维的基质中的高硫蛋白和纤维表皮;大部分片段分子质量集中在25~43 ku之间,这与纤维皮质中间丝的低硫角蛋白有关。UT-1和UT-2的分子质量分布显示出较为分散的条带结构,但条带分布范围有变窄的趋势,大部分片段分子质量集中在31~43 ku之间,这表明从超声波处理的兔毛中提取的角蛋白能够解离成二聚体和三聚体。除此之外,UT-1、UT-2试样的电泳图谱显示,有少量角蛋白片段分子质量集中在97.4 ku左右,且UT-2试样的分子质量在14.4 ku以下减小。当超声波处理时间达到3 h时,角蛋白的分子质量分布连续,且主要集中在大分子质量区域,14 ku左右的分布明显减少。对于UT-4试样,在整个分子质量分布区间基本都有体现,最为明显的区间分布约在16~43 ku之间。由此可知,超声波辅助还原法可提取大分子质量的角蛋白。
2.4 兔毛角蛋白的二级结构分析
2.4.1 化学结构分析

采用拉曼光谱仪测试了兔毛与角蛋白样品中二硫键的变化,如图4(b)所示。可知,兔毛纤维的拉曼光谱中明显观察到位于512 cm-1处S—S键的存在,超声波处理后兔毛纤维在512 cm-1处的吸收峰强度降低,在角蛋白的光谱中S—S键几乎消失,取而代之的是位于1 040 cm-1处的磺酸盐的S—O键[19],这与氨基酸含量分析表现的情况一致。据资料显示512 cm-1处振动峰来自扭-扭-扭(G—G—G)的CC—S—S—CC带构象[20]。在不同的角蛋白样品中,540 cm-1处出现了较为明显的振动,该振动属于反-扭-反(T-G-T)转变。
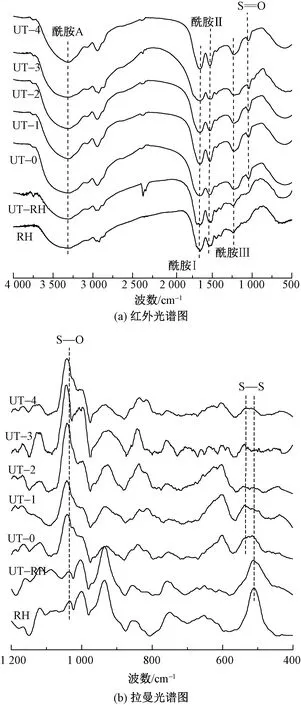
图4 兔毛角蛋白的红外光谱图和拉曼光谱图Fig.4 Infrared spectroscopy (a) and Raman spectroscopy (b) of rabbit hair keratin
红外光谱图中酰胺Ⅰ的吸附带位于1 700~1 600 cm-1之间,对超分子结构特别敏感,该条带通常用于计算α-螺旋和β-折叠结构的含量[21]。为研究超声波作用对兔毛角蛋白二级结构的影响,对红外光谱图进行归一化处理后,用高斯函数解析了酰胺Ⅰ带,其位置、二级结构分配和相关含量百分比如表2所示。可知,兔毛纤维中α-螺旋峰面积较大(位于1 651 cm-1处),无规卷曲峰面积居中,β-折叠峰面积相对较小。随着超声波处理时间的增加,由于二硫键的断裂,蛋白质大分子链被破坏,α-螺旋结构逐渐转换为β-折叠结构和无规卷曲结构。同时,随着超声波处理时间增加,角蛋白的α-螺旋和β-折叠带向较低的波数区域移动,半峰宽度值逐渐降低(即形成更窄的带),这表明角蛋白的二级结构逐渐变得一致[22]。拟合结果还显示,通过超声波处理提取的兔毛角蛋白末端COOH基团含量增加,这与角蛋白提取过程中分子链间氢键、离子键的破坏程度增加有关[18]。

表2 兔毛及其角蛋白的二级结构含量Tab.2 Secondary structure content of rabbit hair and keratin %
2.4.2 结晶结构分析
兔毛及其角蛋白的X射线衍射谱图如图5所示。可知,兔毛和提取的角蛋白均在8.7°和19.12°附近出现典型的蛋白衍射峰,前者归结为α-螺旋结晶结构衍射峰,后者归结为β-折叠结构衍射峰[23]。兔毛纤维主要显示出位于8.7°处的衍射峰,其结构以α-螺旋结晶结构为主。超声波处理的兔毛纤维在8.87°处的衍射峰强度降低,而20.06°对应的衍射峰强度增加,这表明超声波处理过程中,兔毛纤维的部分区域由于超声波作用导致应力集中而使其结构破坏,这与红外光谱酰胺I拟合结果一致。
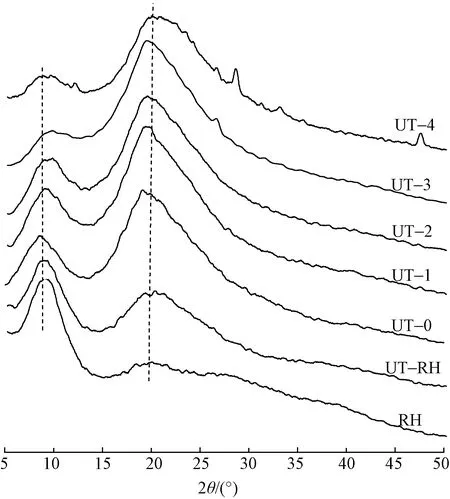
图5 兔毛及其角蛋白的X射线衍射谱图Fig.5 X-ray diffraction for rabbit hair and keratin
与RH相比,提取的角蛋白在8.7°处的衍射峰强度明显减弱,取而代之的是19.12°处的衍射峰强度增加,且衍射峰对应位置向低衍射角方向偏移,由20.06°偏移至19.12°左右。这主要是因为兔毛在超声波和化学试剂的作用下二硫键断裂,进而导致α-螺旋结晶结构破坏;衍射峰位置的减小表明角蛋白内2个相邻晶格晶面间距增加,也说明超声波处理对角蛋白晶体有膨胀作用。处理时间不同,角蛋白的结晶结构也不相同:超声波处理2 h内,提取的角蛋白在8.7°处衍射峰的强度变化相似;当处理时间达到3 h时,该处衍射峰相对强度明显降低,这是因为超声波处理在一定程度上也能破坏纤维的α-螺旋结晶结构。对兔毛纤维与角蛋白的X射线衍射谱图进行去基线后平滑,通过拟合估算了结晶度和晶格间距,结果如表3所示。可知,随着超声波处理时间增加,结晶度明显降低,晶面间距逐渐增大。

表3 结晶度和晶格间距Tab.3 Crystallinity index and lattice spacing
2.5 兔毛角蛋白的三级结构分析
蛋白质的氨基酸组成中,Trp、Tyr以及Phe因具有发色基团而产生荧光。由于Phe在绝大多数实验条件下不被激发,Tyr残基又会将吸收的能量转移给Trp残基,使得Trp残基的荧光增强,所以角蛋白的内源荧光主要来自Trp[24]。Trp属于疏水性氨基酸,微环境的变化对其荧光光谱影响很大,通过色氨酸残基发出的内源荧光光谱可反映出蛋白质结构的变化。兔毛角蛋白的内源荧光光谱如图6所示。可知,提取的兔毛角蛋白具有荧光特征。角蛋白的氨基酸含量测试结果显示色氨酸占0.68%,酪氨酸占4.61%,苯丙氨酸约占3.53%,这3种氨基酸的分子结构存在能够发色的荧光基团——吲哚环和苯基残基。不同超声波处理条件下提取的角蛋白的荧光特征峰位置也不相同。其中,UT-0、UT-1位于 358 nm 处,UT-2和UT-4位于356 nm处,UT-3位于354 nm处,但均处在(356±2) nm范围内,这与Trp的荧光发射光谱相同。随着超声波处理时间的增加,提取的角蛋白荧光发射谱发生很小的蓝移,荧光强度小幅增加。蛋白质荧光光谱受溶剂极性的影响较大,极性环境会影响生色基团的基态和激发态能级,减少激发态的能量,从而引起发射谱的红移,强度降低[25]。由此可知,超声波处理时间越长,提取的角蛋白中芳香族氨基酸分子所处的局部小环境的极性越小,弱于角蛋白分子外部水溶液的极性,其被深埋于角蛋白的内部,被多种非极性氨基酸残基包围。

图6 兔毛角蛋白的内源荧光光谱Fig.6 Endogenous fluorescence spectra of rabbit hair keratin
2.6 兔毛角蛋白粒径和电位分析
蛋白质溶液的稳定性和聚集程度可通过平均粒径和ζ-电位反应出来,超声波处理时间对角蛋白粒径分布的影响如图7(a)所示。可知,随着超声波处理时间的增加,角蛋白的粒径表现出不同程度的偏移,超声波处理时间在2 h以内时,角蛋白粒径分布明显右移,而当超声处理时间增加到3 h后,粒径分布又出现了一定的左移。UT-0、UT-1、UT-2、UT-3和UT-4的平均粒径分别为164.22、179.64、178.81、159.19 和158.53 nm。

图7 兔毛角蛋白的粒径分布与ζ-电位值Fig.7 Particle size(a) and ζ-potential(b) of rabbit hair keratin
不同超声波处理时间下提取的兔毛角蛋白的 ζ-电位如图7(b)所示。可知,超声波处理会使提取的角蛋白的ζ-电位绝对值降低,当处理时间在3 h以内时ζ-电位值基本相近,超声波处理时间增加到4 h时,ζ-电位绝对值降低明显。这说明随着超声波处理时间增加,提取的角蛋白表面净电荷数减少,体系开始变得不稳定。
角蛋白的粒径大小受到大分子链断裂程度、分子间聚集行为以及大分子链伸展情况的综合影响。研究显示超声波处理时间越长,纤维溶解率越大,此时大分子链之间破坏程度增加[26],这可能会导致提取的角蛋白的粒径减小。同时由荧光测试可知,超声波处理时间越长,疏水性的芳香族氨基酸处于蛋白质内部,角蛋白分子链伸展程度越低,所以粒径越小。但角蛋白具有良好的自组装特性,ζ-电位测试结果显示,处理时间增加时提取的角蛋白不稳定,此时角蛋白分子间依靠氢键、离子键等相互集聚,又会使粒径增大,所以角蛋白粒径变化与超声波处理时间变化并不完全相同。
3 结 论
本文详细研究了兔毛角蛋白的化学组成及各级结构,研究结果表明采用超声波辅助还原法提取的角蛋白中氨基酸含量可达88.30%。随着超声波处理时间增加,胱氨酸损失增大,游离巯基呈先增加后减小的趋势,高分子质量蛋白的相对含量增加,分子质量集中分布在31~43 ku之间和97.4 ku以上,有利于角蛋白的再生利用。角蛋白显示出典型的蛋白质结构,随着超声波处理时间的增加,α-螺旋结构逐渐减少,转换为β-折叠和无规卷曲结构,且峰值向较低的波数区域移动,结晶度显著降低。提取的角蛋白具有稳定的三级结构,角蛋白分子不易发生集聚,形成稳定的角蛋白溶液。
