FBXO7与帕金森病的关系研究进展
2022-04-21余娇叶钦勇
余娇 叶钦勇
帕金森病(Parkinson’s disease,PD) 是第二种最常见的老年神经退行性疾病,病理表现为黑质多巴胺能神经元进行性退变和路易小体形成,主要生化改变是纹状体区多巴胺递质水平降低、多巴胺与乙酰胆碱递质失衡,临床表现为运动迟缓、静止性震颤、肌强直、姿势平衡障碍等。PD的平均发病年龄为60~65岁,但大约5%~10%的患者发病年龄在40岁以下,这些早发型PD患者的发病多数与致病基因的突变有关[1]。2008年,Shojaee等[2]首次发现早发型PD的发病与F-box蛋白7(F-box only protein 7,FBXO7)基因突变相关。近年来研究发现FBXO7基因可以通过调控泛素蛋白酶体系统、线粒体自噬及核转录因子κB(nuclear transcription factor-κB,NF-κB)通路等参与PD发病[3]。基于此,本文将围绕FBXO7的结构定位、功能及其在PD发病过程中可能参与的致病机制等展开综述。
1 FBXO7的结构
FBXO7为高等真核生物中的保守蛋白,是F-box蛋白(F-box protein,FBP)家族成员,FBXO7基因位于人类的22q12-q13号染色体上,由9个外显子组成,区域大小约为24.15 kb,其编码一个由522个氨基酸组成的蛋白质[4],蛋白结构包括了 1~78位的泛素样结构域(ubiquitin-like domain,Ubl)、129~169位的细胞周期蛋白激酶6(CDK6)结合位点、 182~325位的FBXO7/PI31结构域、335~378位的F-box基序(F-box domain,FBD)、423~522位的脯氨酸富集区(proline-rich regions,PRR)和466~496位的R(Ar)DP基序[5](图1)。FBXO7包括4种亚型,其中亚型1是该蛋白在大多数组织和培养细胞系中表达最丰富的形式,本文将围绕亚型1的功能及其在PD中的作用机制进行探讨[6-7]。
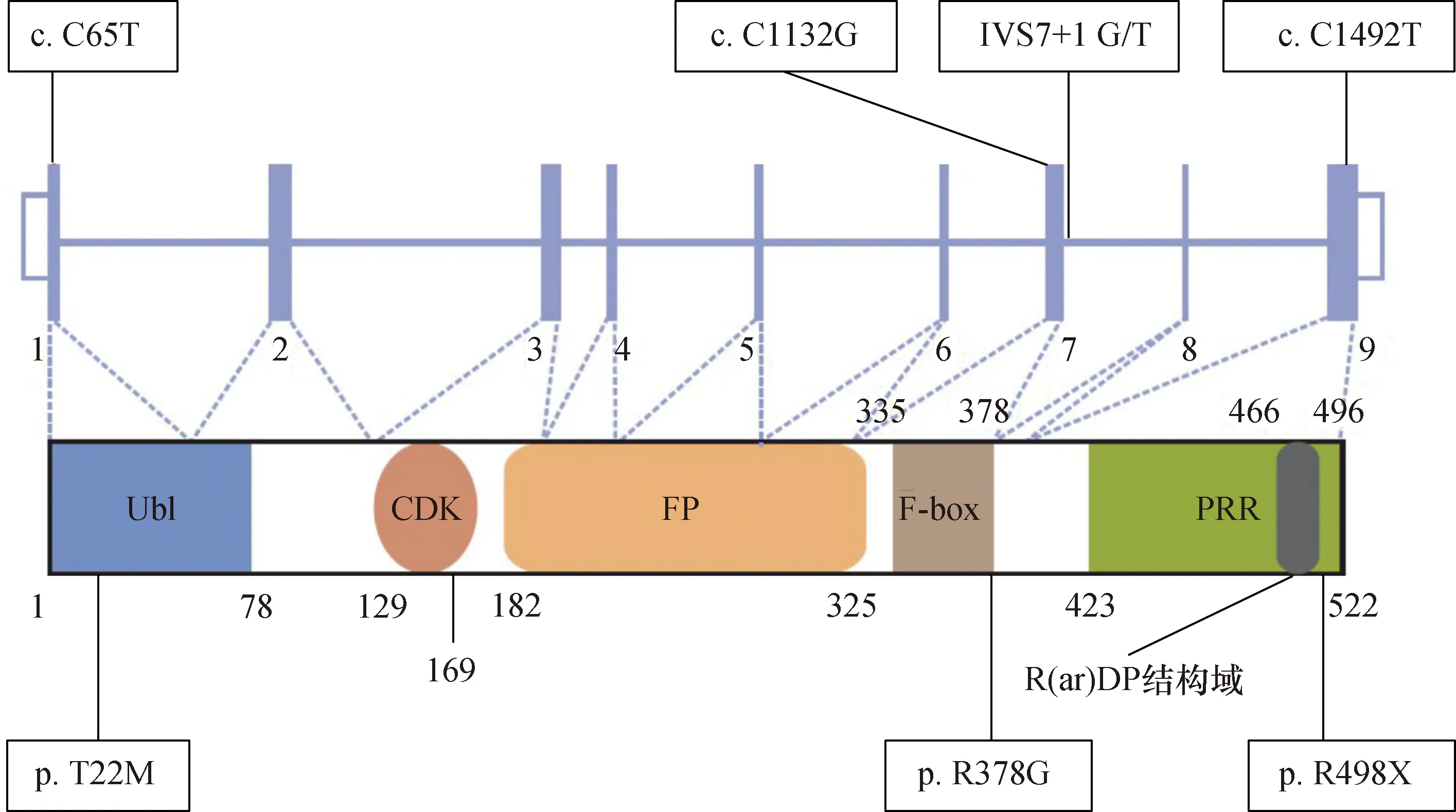
注:FBXO7:F-box蛋白7:Ubl:泛素样结构域;CDK:细胞周期蛋白依赖性蛋白激酶;FP:Fbxo7/PI31相互作用的结构域;F-box:F-box结构域;PRR:富含脯氨酸富集区;N端Ubl中的p.T22M(突变基因的碱基序列为c.C65T),与Parkin直接相互作用;F-box结构域附近的p.R378G(基因突变的碱基序列为c.C1132G)降低了FBXO7形成E3连接酶复合体的能力;位于内含子区域的IVS7+1G/T破坏了FBXO7 mRNA的剪接,干扰mRNA的成熟过程,导致产生功能异常的蛋白质,以及C端的p.R498X(基因突变的碱基序列为c.C1492T;位于蛋白末端附近的一个底物招募结构域) 图 1 FBXO7结构示意图及其突变基因型对应的碱基突变类型[5]
FBXO7突变分布于FBXO7蛋白的多个功能结构域,截至目前共发现7个FBXO7突变基因型[8-9]。其中与PD发生有密切关系的突变有以下5个。(1)纯合错义突变p.R378G位于F-box基序,是其第378位的精氨酸被甘氨酸替代,该突变降低了FBXO7蛋白与s相激酶相关蛋白1(S-phase kinase-associated protein 1,Skp1)的亲和力,破坏了SCFFBXO7(Skp1-Cullin1-F-box)复合物的组装和底物泛素化,极大地影响了细胞的生存和增殖。(2)纯合截断突变p.R498X位于PRR结构域内,是其第498位的精氨酸突变为终止密码子,导致C端缺少脯氨酸结合域,这会削弱SCFFBXO7复合体的E3连接酶活性[10]。(3)剪接位点突变IVS7+1G/T位于内含子区域内,是居间序列7号内含子区域第1个碱基由G变成T,破坏了FBXO7 mRNA的剪接,干扰mRNA的成熟过程,导致功能异常的蛋白质产生[11]。(4)错义突变p.T22M是FBXO7蛋白Ubl结构域内第22位的苏氨酸被蛋氨酸所替代,该突变削弱了FBXO7与帕金蛋白(Parkin)之间的相互作用,损害Parkin到去极化线粒体的易位,而Parkin到去极化线粒体的易位这一过程对启动神经保护性自噬至关重要。(5)纯合突变p.L34R,是N端Ubl域中第34位的亮氨酸被精氨酸替代,与FBXO7蛋白的核定位密切相关[12]。
2 FBXO7的定位
既往研究发现,单体绿荧光双能蛋白(monomeric azami-green geminin,mAG-geminin)和DNA染料Hoechst联合染色后,FBXO7在细胞有丝分裂静止期(quiescent phase,G0)/第一间隙期(gap 1 phase,G1)定位在胞质,当细胞过渡到细胞有丝分裂合成期(synthesis phase,S)/第二间隙期(gap 2 phase,G2)时,FBXO7向细胞核内积聚,这表明FBXO7通常定位于细胞质,在G1/S相变时聚集到细胞核中[13]。FBXO7中存在一个富含亮氨酸的核输出序列(nuclear export sequence,NES),NES负责与核输出蛋白1(CRM1)结合,F-box结构域与Skp1结合。FBD与Skp1的结合阻止了FBXO7与CRM1的接触,这种竞争性结合有助于维持FBXO7的胞质定位[14]。FBXO7在正常人大脑皮层(尤其是额顶叶区域)、纹状体、苍白球、丘脑、黑质、红核、小脑深部核等处表达水平较高,在海马和小脑皮质处表达水平较低。在FBXO7基因突变的PD患者,FBXO7呈低表达。细胞核FBXO7定位需要一个完整的N端,FBXO7相关突变基因p.L34R纯合突变取代了N端泛素样域中的一个高度保守的氨基酸,该氨基酸与FBXO7蛋白的核定位有关。上述FBXO7的细胞定位及区域定位错误与PD的发生均有密切关系。
3 FBXO7的功能及其作用机制
3.1 FBXO7与泛素蛋白酶体系统FBXO7蛋白的F-box结构域通过与Skp1-Cullin复合物结合,形成SCF泛素连接酶复合物,即泛素蛋白酶体系统(ubiquitin-proteasome system,UPS)中的E3连接酶。因此,FBXO7可以参与底物识别和募集以实现泛素化,并随后通过蛋白酶体降解[16]。此外,也有研究显示FBXO7基因敲除小鼠中可以发现蛋白酶体活性降低、早发性运动障碍以及过早死亡[17]。
蛋白酶体抑制剂31(proteasome inhibitor 31,PI31)是蛋白酶体与动力蛋白轻链蛋白(DYNLL1/2)耦联的接头。PI31失活可以抑制轴突中蛋白酶体的运动,破坏突触的蛋白稳定、结构和功能,PI31介导的蛋白酶体轴突运输受损可能是神经退行性疾病发生发展的原因之一[18]。FBXO7蛋白缺失会导致PI31的切割和失活,蛋白酶体活性降低[19]。蛋白酶体的完整性和活性依赖于FBXO7蛋白酶体亚基α2(proteasomal subunit-α2,PSMA2),PSMA2是蛋白酶体核心颗粒的一部分,具有蛋白水解活性。FBXO7基因p.R378G纯合错义突变降低了FBXO7对Skp1的亲和力,导致FBXO7-PSMA2的相互作用受到破坏,使细胞受到蛋白酶体慢性功能障碍的影响,引起异常的泛素化[20],这提示FBXO7的SCF依赖性E3连接酶活性的丧失将是PD发病机制中的一个重要方面。
3.2 FBXO7与线粒体能量代谢及线粒体自噬FBXO7通过其Ubl域直接与Parkin相互作用,促进其被募集到线粒体中,启动线粒体吞噬。位于FBXO7的Ubl结构域中的p.T22M突变消除了其与Parkin的相互作用,从而阻止其补充去极化线粒体,引发线粒体功能障碍[21]。FBXO7缺乏与细胞内NAD+水平降低有关,在线粒体呼吸作用减弱的情况下,线粒体膜电位和ATP含量降低,细胞内活性氧(reactive oxygen species,ROS)产生增加,ROS激活了FBXO7缺陷细胞中聚腺苷二磷酸-核糖聚合酶〔poly (ADP-ribose) polymerase,PARP〕的活性。PARP过度激活导致FBXO7缺陷细胞的线粒体功能障碍,最终导致多巴胺能神经元变性[22]。FBXO7基因p.R498X纯合截断突变可以促进FBXO7从细胞核移位到线粒体并形成有害的FBXO7聚集体,导致线粒体自噬过程的损伤和功能障碍,线粒体清除延迟[23]。
3.3 FBXO7与NF-κB信号通路Spagnol等[24]发现了一种通过E3泛素连接酶FBXO7介导泛表达转录子亚型2(ubiquitously expressed transcript isoform 2,UXT-V2)的蛋白酶体降解来抑制 NF-κB信号通路的新机制,根据其涉及的不同受体,FBXO7在NF-κB信号通路中具有相反的功能。虽然 FBXO7可以正向调节骨形态发生蛋白受体(bone morphogenetic protein receptor, BMPR)-神经营养因子受体相互作用的黑色素瘤抗原基因(neurotrophin receptor-interacting melanoma antigen gene,NRAGE)-转化生长因子β活化激酶(transforming growth factor-Beta activated kinase, TAK1)-转化生长因子β活化激酶结合蛋白(TAK1 binding protein,TAB1)复合物的形成,上调NF-κB活性,但它也通过泛素化肿瘤坏因子(tumor necrosis factor,TNF)受体相关因子2(TNF receptor associated factors 2,TRAF2) 和细胞凋亡抑制蛋白1(cellular inhibitor of apoptosis proteins 1,cIAP-1)负向调节该途径,从而降低受体相互作用蛋白激酶-1 (receptor interacting protein 1,RIP1)泛素化并减弱NF-κB信号传导[24]。此外,错误折叠的α-突触核蛋白积累会激活神经胶质细胞中的几种信号通路,最终导致NF-κB的激活和促炎细胞因子的产生,从而加重神经元退行性改变。
4 FBXO7基因突变与PD的关系
2008年Shojaee等[2]发现了一个患有早发型PD的伊朗大家系,正式命名该病为PARK15,并将FBXO7基因确定为致病基因。据文献报道中国人群中第一例早发型PD,其无义突变E470X可以将翻译产物的第470个氨基酸的密码子从谷氨酸改变为终止密码子,导致蛋白质过早截断产生一种功能失调的FBXO7蛋白;错义突变N51S可以将翻译产物的第51个氨基酸从天冬氨酸改变为丝氨酸,这些均被认为是致病突变[25-26]。PARK15为常染色体隐性遗传,多于40岁以下发病,表现出典型的PD特征(运动迟缓、僵直、静止性震颤和姿势不稳),此外还有痉挛、精细运动障碍和巴宾斯基征(表现为锥体束征),常合并有肌张力障碍、吞咽困难、构音障碍、皮质萎缩和/或认知功能下降等症状。PARK15多数为单侧缓慢或不协调发病,但数年后症状对称,进展相对缓慢,左旋多巴可较好地持续改善帕金森症状,但发生运动并发症更早且更严重[10,27]。
既往研究显示在FBXO7基因敲除小鼠中发现蛋白酶体活性降低、早发性运动障碍以及过早死亡[28-29]。通过免疫组织荧光技术在α-突触核蛋白阳性包涵体(路易体、路易神经突、胶质胞质包体)中检测到FBXO7的免疫反应性,这些发现提示FBXO7在突触核蛋白相关疾病的发病机制中发挥了作用[30]。α-突触核蛋白积累是PD病理学的特征,错误折叠的α-突触核蛋白积累会激活神经胶质细胞中的几种信号通路[31],最终导致NF-κB激活和促炎细胞因子产生,从而加重神经元退行性改变。FBXO7通过FBD与SCF结合介导 UXT-V2 的蛋白酶体降解,抑制 NF-κB信号通路。因此,通过FBXO7抑制NF-κB活性有望成为保护多巴胺能神经元的治疗策略[32]。
综上所述,FBXO7是由522个氨基酸组成、具有多个结构域的蛋白质,它的功能多样性在生命活动中发挥着重要作用。当FBXO7发生突变时会导致常染色体隐性遗传早发型PD,多于40岁前发病,除了典型的PD症状外,还有痉挛、精细运动障碍等锥体束征。PD是多因素、多机制相互作用相互影响的复杂病症,因此从FBXO7分子水平深入研究PD的发病机制,可能为临床治疗提供重要线索。
