一类混配型钌苯配合物的手征光学特性
2022-04-18冯丽霞刘巧玲冯思思侯玉翠王越奎
冯丽霞 刘巧玲 冯思思 侯玉翠 王越奎*,
(1太原师范学院化学系,晋中 030619)
(2山西大学分子科学研究所,化学生物学与分子工程教育部重点实验室,太原 030006)
0 引 言
随着对手性过渡金属配合物在非线性光学[1]、不对称催化[2]和手性分离[3]等领域研究的深入,各类新型手性材料、手性催化剂及手性药物相继问世。其中,三−二亚胺合钌因其特有的发光性能[4]、抗癌活性[5]及作为光敏剂[6]、催化剂[7]等方面的应用而备受关注。目前,基于钌配合物光致手性构型反转[8]、光解离[9]和光致配体取代[10]等反应机理及手性构型构性关系[11]的理论研究很多,而关于钌苯配合物的相关研究尚未见到。
金属苯源自苯分子中CH基团被过渡金属替代后形成的六元金属环形物[12],是一类有机金属化合物。因其不仅表现出有机化合物芳香性的特点,还能调节有机金属反应而受到人们的青睐[13⁃15]。就钌苯配合物而言,早期发现的大都是热力学不稳定的,仅作为有机金属转化过程中的活性中间体[16],故无法研究其多样的化学反应行为。直到Xia等[17]成功合成了室温下稳定存在的钌苯配合物[Ru{CHC(PPh3)CHC(PPh3)CH}Cl2(PPh3)2]Cl后,才为探索其丰富的配位立体化学、亲电/亲核芳香性取代[18]及与环状联烯等配合物转化[19]的反应性创造了条件。此后,Zhang等[20]又将这类配合物的反应性研究拓展到手性领域,发现与菲咯啉(Phen)和L⁃半胱氨酸(L⁃Cys)混配的手性钌苯配合物在乙醇/水混合溶液中能发生金属中心手性构型的动力学反转,且该反转行为受溶液的pH值调控。为理解钌苯体系中pH值调控的手性反转机理,我们拟利用理论计算研究其手征光学性质,以揭示有机金属配合物特有的配位立体化学;探讨八面体金属中心Λ/Δ手性、L⁃半胱氨酸λ/δ折叠及R/S手性碳对电子圆二色(ECD)谱的影响;借助激子手性方法(ECM)分析短波区激子裂分的规律性,并与混配型无机钌配合物比较,阐明二者手征光学特性的异同。
1 计算方法
对比了MPW1K、BB1K与ωB97X⁃D等泛函,最终选定B3LYP及混合基组BS:对Ru使用ECP28MWB赝势基组[21]及含2个f⁃极化函数的增强基组(8s7p6d2f)/[6s5p3d2f][22],其余原子使用 6⁃311G(d)。利用这一基组和泛函,对手性异构体的几何构型进行了优化和频率验证(皆无虚频),并用含时密度泛函TDDFT/B3LYP/BS方法计算了各低能异构体前400个激发态的激发能、振子强度与旋转强度等。所有计算均考虑了溶剂(水)效应,运用Gaussian 09程序包完成。
理论ECD谱的带形采用Gauss函数拟合:
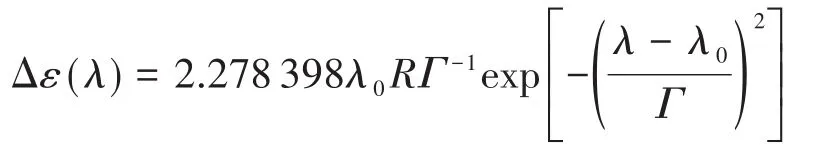
式中,λ0为计算波长(nm);R为旋转强度(DBM);Γ为Δεmax/e处的半带宽,按经验公式Γ=kλ01.5计算[23],当参数k=0.003 85时,模拟ECD与实验谱吻合得最好。
此外,使用ECM[24]对钌苯配合物的激子裂分样式进行了分析。其原理是:如果一个手性分子具有2个或2个以上波长相同或相近的生色团,且这些生色团都具有强的电偶极跃迁,那么在电磁场中,各个生色团间电偶极跃迁矩的偶极−偶极作用,会导致其激发态之间的混合。当这些生色团呈不对称排列时,这种耦合作用会在ECD谱上表现出特定的圆二色吸收样式,即激子裂分。这种由电偶极跃迁产生的耦合作用是一类典型的激子耦合。对于磁偶极允许的跃迁或电偶极允许与磁偶极允许的跃迁之间也能产生耦合,称为非典型的激子耦合。作为一种非经验性的计算方法,对3个配体的跃迁波长和跃迁矩直接采用其TDDFT的计算值。再以此为参数,用自编程序计算配合物的激子耦合波长和旋转强度等,进而模拟ECD谱。因此,相较于TDDFT,ECM法具有原理简单、结果明晰的特点。
2 结果与讨论
2.1 构型参数与相对能
研究体系是与Phen、L⁃Cys混配的3,5⁃双(三苯基膦)钌苯配合物[RuBen(PPh3)2(Phen)(L⁃Cys)]2+,其命名采取“手性符号+名称”的形式,如:Λ(λR)⁃RuBen和Δ(δR)⁃RuBen中的手性符号分别表示配合物中八面体Ru核的Λ/Δ⁃构型、L⁃Cys五元螯合环的λ/δ折叠及R型手性碳。显然,各异构体的手性仅涉及Λ(λR)⁃、Δ(δR)⁃、Λ(δR)⁃和Δ(λR)⁃RuBen 四类。另外,因L⁃Cys上S、N原子均可沿钌苯环的轴向位置(即垂直环平面的位置)与Ru配位。因此,在优化低能异构体时,需同时考虑包含上述4种手性构型在内的8种构象。初步计算显示,所有以N原子做轴向配体的构象异构体均是热力学不稳定的,如图1中Λ(λR)′与Δ(δR)′构象,其能量比最稳定的S配位构象Λ(λR)分别高 37.45、45.70 kJ·mol−1。该结果与实验事实完全吻合[20],故以下仅讨论S原子发生轴向配位的手性构型。
图1显示了B3LYP/BS水平优化的水溶液中4种手性构型Λ(λR)⁃、Δ(δR)⁃、Λ(δR)⁃和Δ(λR)⁃RuBen的几何及类似物Crystal的晶体结构[25],相应主要构型参数列于表1。为便于讨论,文中各异构体的原子编号及坐标系的规定均以图1示例中Λ(λR)为参考。因当前体系的晶体数据未见报道,故下面选类似体系的实验数据比较。总体而言,理论计算的配位键长、键角与二面角均与类似物晶体数据吻合。其中Ru—N1键(约0.213 9 nm)最接近实验键长0.218 7 nm,而Ru—N2与Ru—N3键的偏差或许源于2类体系不同的配位环境。此外,各手性异构体的中心Ru原子均具有轻微扭曲的八面体结构,N1—Ru—N2、N3—Ru—S键角分别约为75.08°、81.28°,均偏离理想的90°。如预期所料,钌苯环具有一定的芳香性特点:整个六元环几乎共面,且环内C—C键长趋于平均化(约0.14 nm),这保证了环平面离域大π键的形成。以Λ(λR)⁃RuBen为例,C1—C2、C2—C3、C3—C4与C4—C5的键长十分接近,最大偏差仅为0.000 8 nm,2个共价键Ru—C1与Ru—C5的键长为0.191 3、0.192 0 nm,与类似物Ru—C键长0.191 6 nm非常吻合。二面角D(C1—C2—C4—C5)、D(C2—C1—C5—Ru)与D(C1—C2—C4—C3)分别为 1.02°、170.45°与−176.96°;此外金属环中6个内角之和718.4°与六边形内角和720°几近相等的事实均说明钌苯环准共面。该共面性保证了Ru原子d轨道与环内C原子pz轨道的重叠,便于形成dp⁃π型离域键,详见Kohn⁃Sham(KS)轨道分布。这一结构特点合理解释了锇苯[13]、钌苯[18]等有机金属配合物易发生亲电/亲核芳香性取代的实验事实。
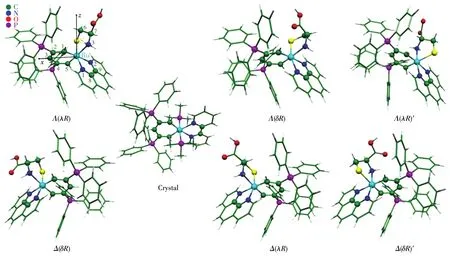
图1 优化的手性异构体、规定的原子编号与坐标系Fig.1 Optimized chiral isomers,the atom numbering for part of atoms and the coordinate system
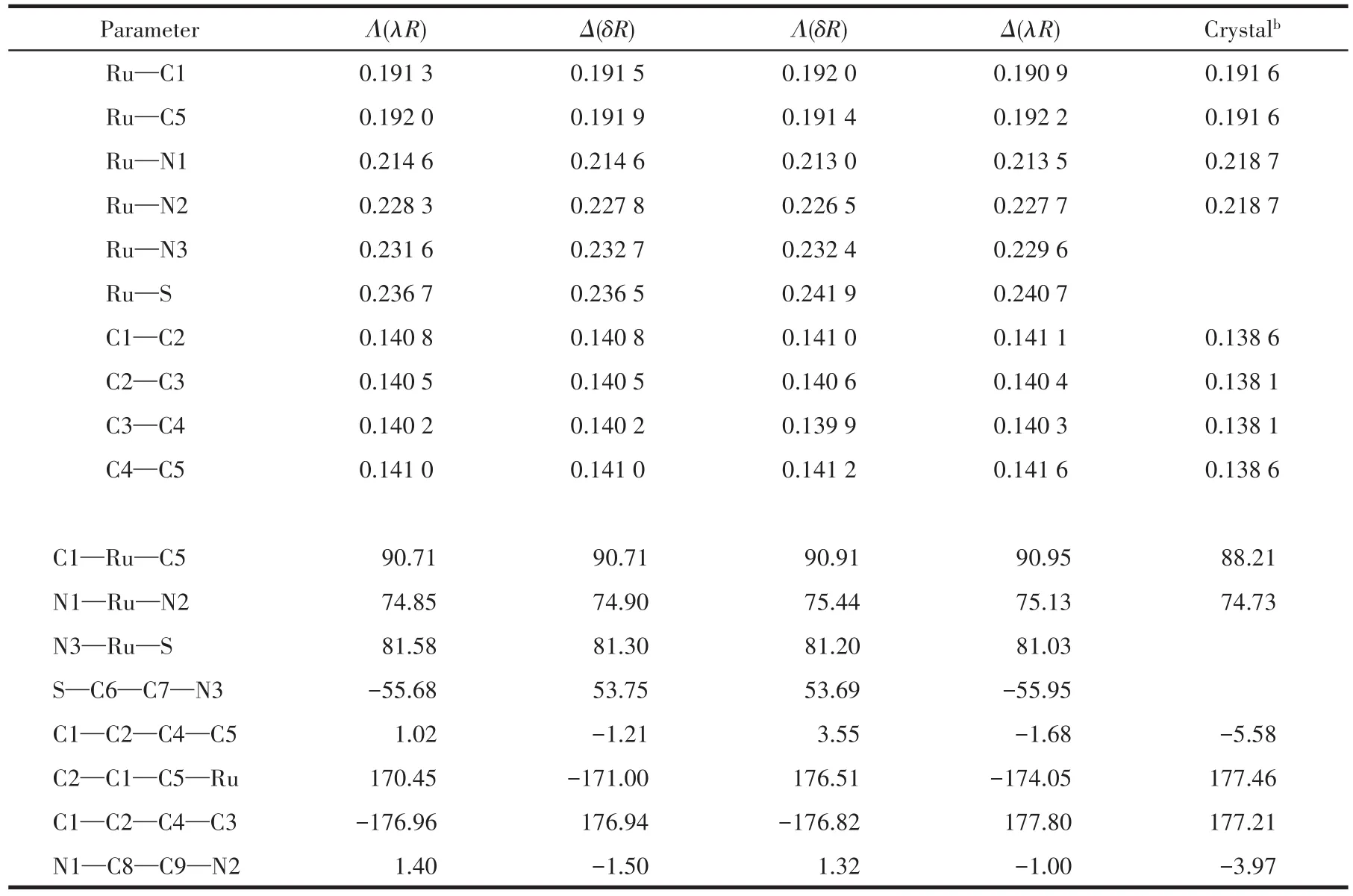
表1 低能异构体的主要构型参数与相关晶体结构Table 1 Main geometrical parameters of low-energy isomers and the relevant crystal structurea
以Λ(λR)为参考能量零点,得到水溶液中手性异构体Λ(λR)⁃、Δ(δR)⁃、Λ(δR)⁃和Δ(λR)⁃RuBen经零点能(ZPE)校正的相对能量分别为0.00、0.53、15.47和7.59 kJ·mol−1。就Λ/Δ手性构型而言,前一组Λ(λR)与Δ(δR)的能量较之后一组Λ(δR)与Δ(λR)更低。低能异构体中,Δ(δR)的能量仅比Λ(λR)高0.53 kJ·mol−1。
2.2 DFT能级与KS轨道
非对映异构体Λ(λR)⁃与Δ(δR)⁃RuBen、Λ(δR)⁃与Δ(λR)⁃RuBen的DFT能级图绘于图2。因图中各异构体的轨道数目与种类大致相同,且KS轨道分布类似,图3仅展示最稳定构型Λ(λR)的主要KS轨道。以钌苯环内Ru⁃C3轴为x轴,通过Ru原子垂直于环平面的方向为z轴(几乎通过Ru—S键),与x、z垂直的方向为y轴,建立直角坐标系(图1),并详细分析图2与图3可知,钌苯配合物的占据轨道与空轨道存在较大能隙。以Λ(λR)⁃RuBen为例,前4个最高占据分子轨道(HOMO)中,除239号轨道为Ru中心的轨道外,其余237、238和240均为Ru(Ⅱ)的d轨道与环内C原子pz轨道重叠而成的离域dp⁃π键,简记为dp⁃π(RuBen)(图 2)或π[RuBen(d)](图 3),后者可显示参与成键的d轨道类型。除此之外,上述轨道连同最低未占据分子轨道(LUMO),即237~241中均含有大比重的S原子孤对电子占据的p轨道贡献,记为nS。紧邻的235、236轨道是菲咯啉配体的π成键轨道,记为π(Phen)。后续密集的222~234能带源自PPh3中各苯环(Ph)的π成键,即π(Ph)。其余218~221的占据轨道分别为d轨道参与形成的dp⁃π、dp⁃σ型成键MO,其中,218中尚含有L⁃Cys中—COOH上2个O原子孤对电子占据的p轨道成分,记为nO;dp⁃σ源自Ru(Ⅱ)的dxy与C、N的p轨道形成的σ(Ru—C,Ru—N)键。反键轨道的分布与成键轨道类似,分别为241的dp⁃π*(RuBen)和242、243的π*(Phen),除251也属π*(Phen)外,244~256均归属于π*(Ph)。在所有高能空轨道中,258与259表现为Ru(Ⅱ)的dz2轨道参与形成的 6个反键dp⁃σ*(Ru—S、3Ru—N、2Ru—C)及L−Cys上π*CCOO轨道;261~265大多为Ru原子s或p轨道形成的Rydberg(Ry)态。

图 2 Λ(λR)⁃RuBen(a)、Δ(δR)⁃RuBen(b)、Λ(δR)⁃RuBen(c)和 Δ(λR)⁃RuBen(d)的DFT 能级图Fig.2 DFT energy levels of Λ(λR)⁃RuBen(a),Δ(δR)⁃RuBen(b),Λ(δR)⁃RuBen(c),and Δ(λR)⁃RuBen(d)
需特别指出的是,钌苯配合物金属中心的d轨道参与了芳香性MO的形成。图3中199、237、238号占据轨道及241、257、262号空轨道,与苯分子中具有芳香性的6个π型MO非常相似:199轨道的面包圈构型对应苯中HOMO⁃2轨道的超环面电子云分布;237与238、241与257轨道分别对应苯中含1个、2个节面的二重简并HOMO、LUMO;262轨道对应苯的LUMO+2。以上成键特点说明钌苯环具有芳香性[15,19]。
2.3 ECD谱与跃迁特性
2.3.1 理论谱与实验谱比较
为明确指认钌苯配合物实验ECD谱的电子跃迁行为,借助TDDFT/B3LYP/BS方法计算了水溶液中Λ(λR)、Δ(δR)、Λ(δR)与Δ(λR)的前200个激发态;并额外补算了低能构象Λ(λR)与Δ(δR)的另200个激发态(共计400个),以预测配合物在远紫外区(λ<230 nm)的跃迁属性。计算谱与实验谱[20]对照如图4所示,其中绿色短棒代表跃迁位置与相对强度;红色ECD曲线旁的数据对应主要吸收带的峰值波长λmax与摩尔消光系数Δεmax。因实验谱与计算谱的纵坐标单位不同,仅能对比两谱的相对强度。在250~600 nm的实测波长范围内,2个低能异构体Λ(λR)与Δ(δR)的ECD曲线与实验谱[20]的带型分布基本吻合:尽管其谱带位置有所红移或蓝移,但峰谷数目与符号均完全相同。如Λ(λR)中3个正、负吸收带的相对强度与实验谱中的Λ⁃手性构型一致。
2.3.2 手征单元对ECD谱的贡献
纵向对比图 4 中Λ(λR)与Δ(δR)、Λ(δR)与 Δ(λR)的ECD曲线发现,2组谱图几乎互为镜像,说明R/S手性碳对ECD谱的贡献很小。横向比较Λ(λR)与Λ(δR)计算谱,特别是二者与相应Λ⁃构型实验谱的吻合程度发现,虽然Λ(δR)构型中峰谷数目及符号也与实验谱一致,但其长波区400 nm以上谱带的红移现象非常严重,且ECD谱的相对强度与实验谱完全不同,如实验谱中位于446.0 nm附近的负峰强度明显高于 361.4 nm处,而Λ(δR)中相应位置491.2、348.2 nm处的负峰强度则完全相反,类似特点见图4d~4f。显然,螯合环小手性λ与δ的折叠方式会影响ECD谱的相对强度。交叉对比Λ(λR)与Δ(λR)(图 4b、4f)或Λ(δR)与Δ(δR)(图4c、4e)的 ECD分布,2组曲线不仅出峰位置不同,且Cotton效应的符号与带形几近相反,说明金属中心Λ与Δ的手性构型才是支配ECD曲线分布的主因。显然,当前体系中,各手征结构单元对ECD谱的贡献顺序为金属中心Λ/Δ⁃构型>螯合环的λ/δ折叠>R/S手性碳。

图4 Λ⁃构型 (a)和Δ⁃构型 (d)RuBen的实验ECD谱;Λ(λR)⁃RuBen(b)、Λ(δR)⁃RuBen(c)、Δ(δR)⁃RuBen(e)和 Δ(λR)⁃RuBen(f)的计算ECD 谱Fig.4 Observed ECD spectra of Λ⁃configuration(a)and Δ⁃configuration(d)RuBen;Calculated ECD spectra of Λ(λR)⁃RuBen(b),Λ(δR)⁃RuBen(c),Δ(δR)⁃RuBen(e),and Δ(λR)⁃RuBen(f)
2.3.3 主要跃迁性质
为明确解析并指认ECD谱,表2列出异构体Λ(λR)⁃RuBen在200 nm以上的主要光学跃迁,包括跃迁波长λ、振子强度f、旋转强度R、电(μ)与磁(m)偶极跃迁矩及二者夹角θ等。
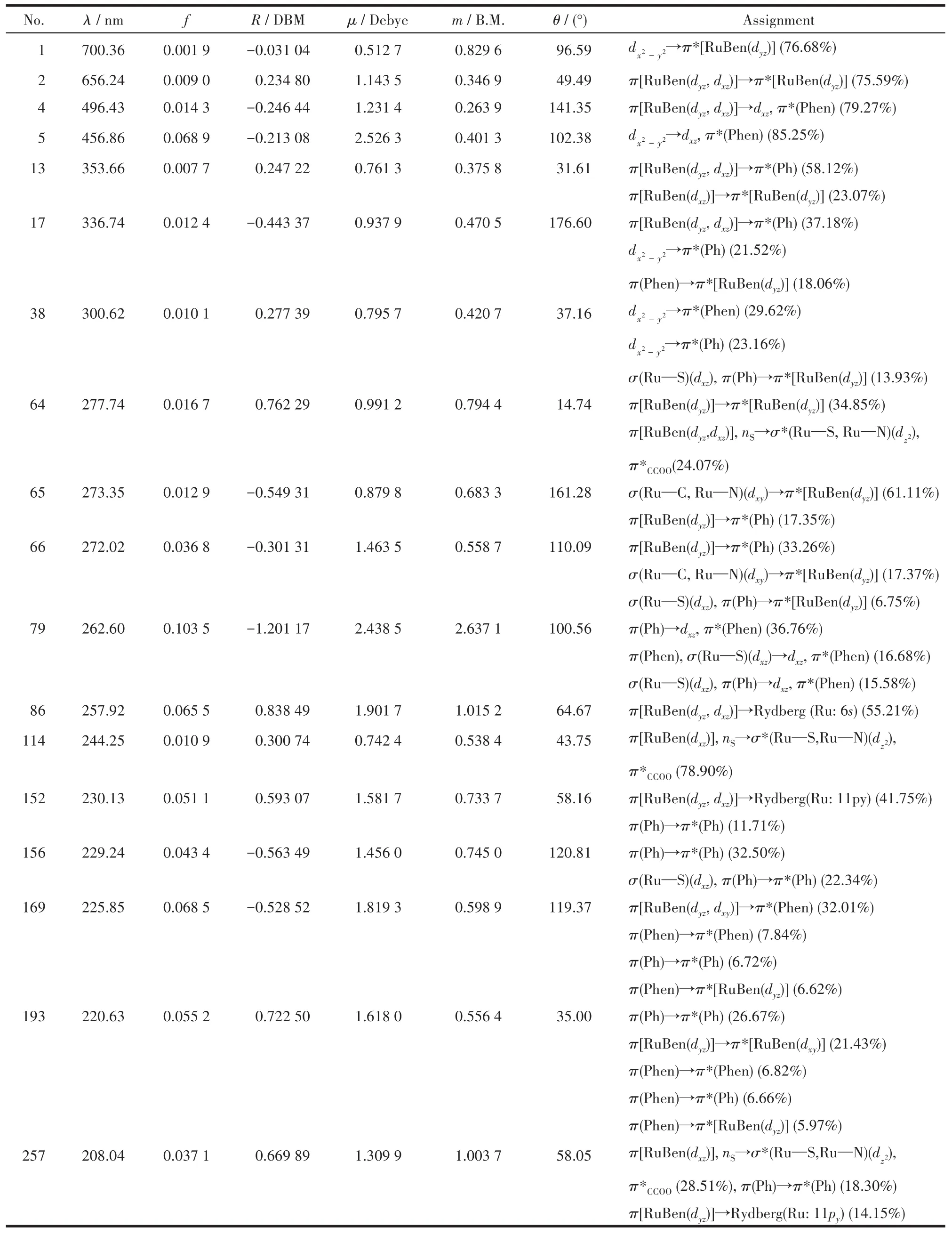
表2 Λ(λR)的激发态编号(No.)、波长(λ)、振子(f)与旋转强度(R)、电(μ)与磁(m)偶极跃迁矩及二者夹角(θ)和主要跃迁Table 2 Number of excited states(No.),excitation wavelengths(λ),oscillator(f)and rotational strengths(R),electric(μ)and magnetic(m)dipole transition moments and the angle(θ)between them,and the dominant transition assignments of Λ(λR)
对异构体Λ(λR),实验ECD谱长波区(约600 nm)第一个正带主要源自钌苯环内部的dp⁃πRuBen→dp⁃π*RuBen跃迁,并混有部分Ru中心的d→d成分。紧邻的宽负带(400~530 nm)含钌苯与菲咯啉配体间dp⁃πRuBen→π*Phen及金属−配体d→π*Phen荷移跃迁。实验谱中392、361 nm处的弱正与弱负带皆由钌苯环与环外围取代基苯的dp⁃πRuBen→π*Ph跃迁主导,前者含πRuBen→π*RuBen,后者含πPhen→π*RuBen,对应计算谱中361、334 nm处的峰与谷。短波区340 nm以下一正一负的强吸收带呈典型的激子裂分样式,源于2个平面配体RuBen与Phen的跃迁耦合,表现为金属−配体d→π*Phen、钌苯环内π→π*与σ→π*及Phen的π→π*等,与295、265 nm处的计算谱对应。此外,计算谱中多出的一正一负2个弱峰揭示了配合物短波区(200~250 nm)的跃迁特性:234 nm处的正峰涉及πRuBen到高能Rydberg态的激发,同时含L⁃Cys配体内部的nS→π*CCOO跃迁;215 nm处的负峰涉及πPhen→π*RuBen跃迁。事实上,计算谱中330 nm以下的2对正负分布曲线均具有激子耦合特性,相关讨论详见下一部分。分析显示,当λ<230 nm时,来自远程取代基Ph内部的π→π*跃迁尤为突出,说明PPh3基团的跃迁主要发生在远紫外区。非对映异构体Δ(δR)⁃RuBen的跃迁行为与此类似。
2.3.4 激子裂分特性
为了直观地理解图4计算谱中短波区复杂的谱带样式,我们又用ECM进行了分析。其中的有关参数,即3个配体的跃迁波长及其电偶极和磁偶极跃迁矩,直接采用计算值(泛函和基组同前)以免引入人为参数,对配体间的作用则取偶极−偶极近似,结果如图5所示。分析表明,其中250 nm左右的一对强吸收带,主要起源于RuBen和Phen两个平面配体π→π*跃迁之间的耦合,表现出典型的激子裂分样式[26]。而210 nm附近的一对弱吸收带,则起源于L⁃Cys的内禀磁偶极跃迁nS→π*和Phen的π→π*诱导磁偶极的耦合,表现为非典型的激子裂分。由于配体的跃迁能直接采用了其计算值,故计算谱发生蓝移。若将计算谱整体红移0.48 eV,则可与前述图4b、4e短波区的谱带很好地匹配。以上2对双信号激子耦合可作为指认类似手性配合物绝对构型的判据,即Λ⁃构型的强带与弱带均具有正手性激子裂分样式,Δ⁃构型具有负手性激子裂分样式。
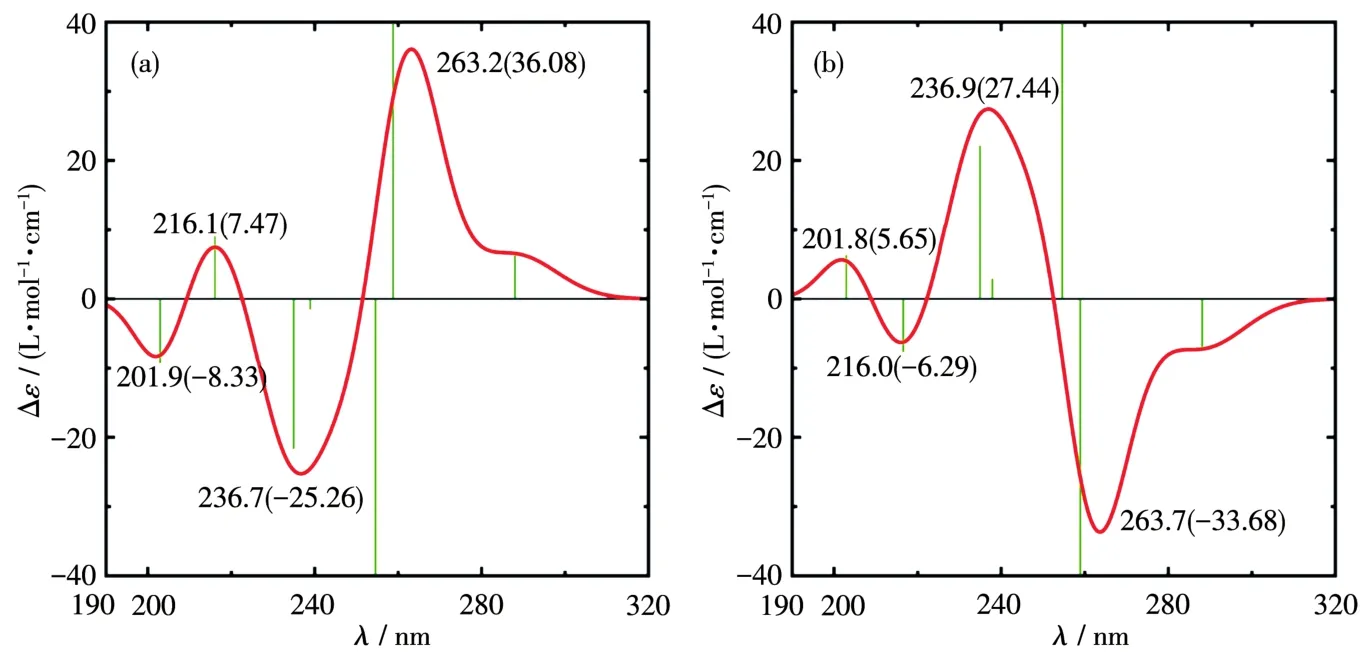
图5 ECM计算的Λ(λR)⁃RuBen(a)与Δ(δR)⁃RuBen(b)激子裂分Fig.5 Exciton splitting of Λ(λR)⁃RuBen(a)and Δ(δR)⁃RuBen(b)calculated by ECM
2.3.5 有机钌苯与无机钌配合物ECD谱比较
图 6 展示了与L⁃丝氨酸(L⁃Ser)、Phen混配的无机钌配合物[Ru(Phen)2(L⁃Ser)]+的计算ECD谱[8]。与本工作研究的混配型钌苯配合物相比,尽管2类体系的配体组成类似,但其ECD曲线的出峰位置、强度及带型分布均不同。
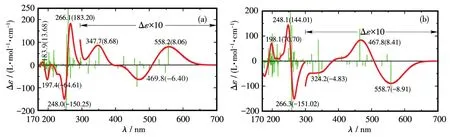
图6 混配型无机钌配合物Λ(δS)⁃[Ru(Phen)2(L⁃Ser)]+(a)与Δ(δS)⁃[Ru(Phen)2(L⁃Ser)]+(b)的计算ECD谱[8]Fig.6 Calculated ECD spectra of mixed⁃ligand inorganic ruthenium complexes Λ(δS)⁃[Ru(Phen)2(L⁃Ser)]+(a)and Δ(δS)⁃[Ru(Phen)2(L⁃Ser)]+(b)[8]
无机钌配合物的计算ECD谱中,除350 nm附近的吸收带属于掺有明显Ry特性的d→dp⁃σ*(Ru—N)金属中心跃迁外,300 nm以上的激发大多为d→π*phen金属−配体荷移(MLCT)跃迁。短波区300 nm以下一强一弱的2对谱带同样具有典型与非典型的激子耦合特征,前者起源于2个Phen的π→π*跃迁,而后者源于L⁃Ser磁偶极跃迁n→π*与Phen的π→π*诱导磁偶极的耦合。其中强带对Λ⁃构型表现为正的手性激子裂分,而弱带则为相反的负激子裂分。该特性不同于钌苯体系,可作为区分无机与有机钌配合物绝对构型的参考判据。
钌苯体系的计算ECD谱中,除450 nm附近的吸收带具有部分类似于无机体系的d→π*MLCT特征外,长波区340 nm以上的跃迁均由π→π*主导,表现为钌苯环内部、其与菲咯啉间及与外围苯分子的π→π*。短波区340 nm以下2对激子中,弱带的裂分样式与无机体系相反,且在小于230 nm的更短波区存在大量来自钌苯环外围取代基Ph的π→π*跃迁。
3 结论
基于上述对混配型有机钌苯配合物的电子构型、ECD谱带归属及激发态性质等的分析,主要结论如下:
(1)计算ECD谱与实验谱的峰形、峰谷数目、符号及相对强度吻合。
(2)各手征结构单元中,八面体钌核的Λ/Δ⁃构型决定了ECD曲线的分布,λ/δ折叠仅影响吸收带的相对强度,而R/S手性碳的贡献极小。
(3)有机钌苯配合物的手征光学性质与混配型无机钌配合物不同。前者长波区的跃迁由π→π*主导,后者则为d→π*型金属−配体荷移跃迁。两者在短波区均涉及一强一弱2对激子耦合带,但其中弱带呈现的激子耦合作用相反。
