Au@g⁃C3N4支架的制备及其光催化性能
2022-04-18刘苗苗吴禧航黎水平汪园园胡学敏张小娟
林 青 刘苗苗 吴禧航 黎水平 汪园园 胡学敏 王 威 张小娟
(1金陵科技学院材料工程学院,南京 211169)
(2扬州大学建筑工程学院,扬州 225127)
0 引 言
光催化氧化技术被认为是降解水体中有机污染物的最经济有效的方法,其核心依靠的是高效、稳定的半导体光催化材料[1]。石墨相氮化碳(g⁃C3N4)是一种非金属n型半导体光催化材料,具有化学稳定性高、结构与性能易于调控、成本低、易制备和无污染等优点[2⁃3]。与二氧化钛(TiO2)、氧化锌(ZnO)等光催化材料相比[1],g⁃C3N4结构中C和N原子以sp2杂化形成高度离域的π共轭系,禁带宽度约为2.65 eV,吸收边约为460 nm,具有良好的可见光响应特性[4⁃5]。利用可见光催化氧化降解水中有机污染物,具有能耗低、效率高和无二次污染等优点,在环境污染物降解处理领域具有良好的应用前景[6⁃7]。但块状g⁃C3N4存在比表面面积小、光生载流子复合过快、可见光吸收范围窄、量子效率低等缺点,其光催化活性有限,光催化降解有机物效率不太理想[8]。因此需要通过多种方法和手段来提高g⁃C3N4的光催化性能,如元素掺杂[9]、构建异质结[10]、形貌调控[4]、贵金属修饰[11]等。g⁃C3N4一般由三聚氰胺、尿素等前驱体经过热聚合法制得[4],此类前驱体聚合后黏度增加,可通过微波发泡法构建出三维支架结构,热聚合后可赋予g⁃C3N4独特的三维支架结构,这将是一项构建高比表面积g⁃C3N4的有益探索。此外,金属纳米粒子(Au、Ag、Cu等)具有表面等离子体效应[11],能有效拓宽催化剂的可见光吸收范围[12],在g⁃C3N4支架表面沉积Au可形成金属−半导体异质结,提高电荷迁移效率,抑制光生电子−空穴对的复合[11,13],可有效提高g⁃C3N4的光催化性能。
我们通过磁控溅射法在微波发泡法构建的三聚氰胺树脂(melamine⁃formaldehyde,MF)支架上沉积Au纳米粒子,热聚合后成功制备了负载Au的g⁃C3N4支架(Au@g⁃C3N4scaffold),采用多种表征手段对其结构和性能进行了研究,并考察了Au@g⁃C3N4支架光催化降解有机污染物的性能。
1 实验部分
1.1 材料制备
三聚氰胺(C3H6N6)、甲醛(HCHO,37%)溶液、尿素、吐温⁃80、正戊烷、乙酸、偶氮二异丁腈、氢氧化钠、罗丹明B(RhB)均购自国药集团化学试剂有限公司,Au靶(99.99%,Φ50 mm×1 mm)购自北京金源新材料有限公司。
采用微波发泡法制备MF支架[14⁃15]。先用10%氢氧化钠溶液将100 mL甲醛溶液的pH值调节至9.0,再加入56.0 g三聚氰胺(甲醛与三聚氰胺的物质的量之比为3∶1),搅拌均匀后,升温至90℃,反应3 h后得到MF溶液;待冷却至室温后,依次添加改性剂(尿素,质量分数10%)、乳化剂(吐温⁃80,质量分数3.0%)、发泡剂(正戊烷,质量分数 10.0%)、开孔剂(偶氮二异丁腈,质量分数5.0%)、固化剂(乙酸,质量分数8.0%),搅拌均匀后平铺于硅胶模具中,最后置于90℃的微波反应器中(FCMCR⁃3SX,瑞科,中国)发泡3 min;取出后,200℃下固化定型得到MF支架。采用磁控溅射法在MF支架上溅射Au(图1):将2.0 cm×2.0 cm×0.2 cm的MF支架置于磁控溅射仪(ACS⁃4000⁃C4,ULVAC,日本)内,双面溅射Au,时间为90 s,得到负载Au的MF(Au@MF)支架。将Au@MF和MF支架置入管式炉(OTF⁃1200X⁃Ⅱ,科晶)内,通入氮气保护,以5℃min−1的速率加热到550℃,保温2 h,冷却到室温,得到Au@g⁃C3N4和g⁃C3N4支架。
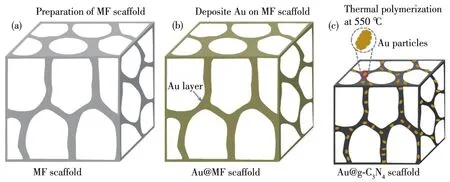
图1 制备Au@g⁃C3N4支架的示意图Fig.1 Schematic illustrations of the preparation of Au@g⁃C3N4scaffold
1.2 材料的测试与表征
采用综合热分析仪(Netzsch Sta449C,NETZSCH,Germany),以10 ℃·min−1的升温速率从室温加热到800℃,对样品进行差示扫描量热(TG⁃DSC)分析。用ARL XTRA型X射线衍射仪(XRD,Thermo Elec⁃tron,America,U=4 kV,I=40 mA,CuKα辐射,波长0.154 2 nm,扫描速度 10(°)·min−1,扫描范围 10°~70°)对样品进行晶相分析。采用傅里叶变换红外光谱仪(FTIR,Nexus 670,Nicolet,America)记录样品的红外吸收光谱,采用KBr压片法制样,扫描次数为32,分辨率为 2 cm−1,扫描范围为 400~4 000 cm−1。采用拉曼光谱仪(HR800,Horiba,Japan)记录样品的Raman谱图,激发光源波长为780 nm。用场发射扫描电镜(SEM,Su8010,Hitachi,Japan,工作电压为3 kV)观察样品表面和断面形貌。采用固体紫外可见漫反射光谱仪(UV⁃Vis DRS,Evolution 500,Thermo,American)测试样品的光吸收性能,以硫酸钡为标准物质,采谱范围为200~800 nm。采用荧光光谱仪(F⁃7000,Hitachi,Japan)测试样品在室温下的光致发光(PL)谱图,激发波长为350 nm。采用X射线光电子能谱(XPS,ESCALAB 250,Thermo,American)分析样品的元素组成和化学态。依据GB/T528—2009,采用电子万能力学实验机(Instron 6022,Instron,American)测试样品应力−应变曲线,拉伸速率为10 mm·min−1。采用比表面积分析仪(ASAP2020M,Micromeritics,American)测试支架的吸附等温线,根据N2吸附−脱附等温线计算得到Brunauer⁃Emmett⁃Teller(BET)比表面积。采用全自动真密度测定仪(G⁃DenPyc⁃2900,金埃谱,北京)测试样品的密度,进而得到开孔率。
1.3 光电化学测试
采用电化学工作站(CHI660E,上海辰华)对样品进行光电化学测试。将样品夹在铂金夹具上作为工作电极,Ag/AgCl电极为参比电极,Pt电极为对电极。光电流测试的电解质溶液为0.5 mol·L−1Na2SO4,工作电压为−0.2 V,使用光源为300 W氙灯(λ≥420 nm),光源开闭间隔20 s进行光电流信号采集。电化学阻抗谱(electrochemical impedance spec⁃troscopy,EIS)在开路电压下,0.1~105kHz频率范围内测量。
1.4 光催化性能测试
通过测试样品在可见光条件下催化降解RhB溶液的能力来评估材料的光催化性能。以无光催化剂的纯RhB溶液(20 mg·L−1)为参照,将样品置入RhB溶液后,先在黑暗处暗吸附60 min,使其达到吸附−脱附平衡,开启氙灯(300 W)对溶液进行照射,每隔30 min取一次样,用TU⁃1901型紫外可见分光光度计测定溶液中RhB的含量,绘制降解曲线。每照射210 min为1次循环,每次循环结束,去除催化剂支架,清洗干燥后,再重新测试,反复6次循环测试后,绘制其循环催化降解曲线。
2 结果与讨论
2.1 形貌分析
图2为MF和Au@MF支架实物照片和SEM照片,由图可知,MF支架纯度高,外表为白色(图2a),MF支架内完全以纤维丝相连,纤维丝直径为5~10 μm(图2b),纤维丝之间形成牢固的结点,呈现三维立体网络结构,孔均敞开连接,孔径大于150 μm。这主要由于MF具有较高黏度,在发泡剂的作用下,内部能形成大量的气泡,微波加热使其体积急剧增大,在开孔剂抑制下,气泡达到一定大小后因表面能过大而破裂[14⁃15],破裂后的气泡形成了连通孔道结构,树脂固化后即形成连通的孔道结构,从而成功制备得到MF支架(图2b)。由于磁控溅射仪溅射沉积于MF支架上的Au粒子尺寸较小,纳米小尺寸效应导致Au@MF支架表面呈现灰黑色(图2c),未呈现块状Au的金黄色。由于MF支架开孔率高,对溅射的Au纳米粒子的阻碍小。断面SEM照片(图2e)表明Au在MF支架内的溅射深度可达1.5 mm,通过双面溅射法,可以使厚度为2 mm的MF支架内部均匀地沉积Au颗粒(图2f),溅射90 s后,Au@MF支架表面的Au层厚度可达14 nm(图2f)。MF和Au@MF支架经550℃热缩合后,分别得到g⁃C3N4和Au@g⁃C3N4支架,体积均缩小约40%,两者外观无显著差异,均呈灰色(图3a)。热聚合后,与MF支架相比,g⁃C3N4和 Au@g⁃C3N4支架的纤维丝直径均缩小至 5 μm以下,孔径缩小至100 μm左右;与Au@g⁃C3N4支架相比,g⁃C3N4支架的纤维丝结点热收缩严重,呈圆形(图3c);由于 Au层的保护,Au@g⁃C3N4支架内纤维丝形态保持良好(图3e)。热聚合过程中,Au@g⁃C3N4支架表面的Au层发生了熔化,由纳米Au层转化为彼此相互独立的Au颗粒(图3f)。

图2 MF和Au@MF支架实物照片 (a、c、f)和SEM照片 (b、e、g)Fig.2 Photos(a,c,f)and SEM images(b,e,g)of MF and Au@MF scaffolds

图3 g⁃C3N4和Au@g⁃C3N4支架实物照片 (a、c、f)和SEM照片 (b、e、g)Fig.3 Photos(a,c,f)and SEM images(b,e,g)of g⁃C3N4and Au@g⁃C3N4scaffolds
Au@g⁃C3N4支架断口SEM照片如图4所示,从图中可看出,Au@g⁃C3N4支架基体内部存在大量的大孔,因在支架纤维丝内部,所以仅能从支架纤维丝断裂面观察到。这主要由于发泡过程中部分气泡残存在树脂内[14⁃15],最终成为 Au@g⁃C3N4支架基体内部的闭孔。三维网络结构和基体内部大孔可赋予Au@g⁃C3N4支架巨大的比表面积,Au@g⁃C3N4支架对有机染料可能具有吸附作用[16]。

图4 Au@g⁃C3N4支架断口SEM照片Fig.4 SEM images of Au@g⁃C3N4scaffold fractures
2.2 TG⁃DSC分析
图5为MF和Au@MF支架的TG⁃DSC曲线,三聚氰胺的热分解吸热范围一般在297~390℃[2],由于MF是三聚氰胺等的聚合物,其热稳定性高于纯三聚氰胺,MF和Au@MF支架分解的吸热峰(398℃)相对较高。SEM照片表明Au@MF支架表面的Au层在热聚合过程中发生了熔化再结晶(图3f),因此Au@MF支架在415℃处出现的吸热峰为纳米Au的熔化吸热峰[17]。经550℃热聚合后,MF和Au@MF支架质量分别降低至38%和42%,这是其发生体积缩小的主要原因,据此也可推算出Au@g⁃C3N4支架上Au的负载量为6%。由于支架具有三维结构,Au@g⁃C3N4支架的质量降低率(78%,图 5)大于其体积减小率(40%,图3a)。

图5 MF和Au@MF支架的TG⁃DSC曲线Fig.5 TG⁃DSC curves of MF and Au@MF scaffolds
2.3 结构分析
图 6a为 g⁃C3N4和 Au@g⁃C3N4支架的 XRD 图,27.6°处对应的是 g⁃C3N4的(002)晶面衍射峰[18],归因于芳香族单元的层间堆积,表明其具有类石墨层状结构;该峰相对较宽,衍射强度较弱,说明g⁃C3N4和Au@g⁃C3N4支架的层间堆积有序度较低,晶粒尺寸较小[19]。13.3°处对应的是 g⁃C3N4的(100)晶面衍射峰[20],归因于其面内3⁃s⁃三嗪单元的有序排列。虽然MF的热分解温度高于纯三聚氰胺(图5)[2],但是XRD图表明MF和Au@MF支架在550℃均能聚合形成g⁃C3N4,550 ℃是 MF 聚合形成 g⁃C3N4的合适温度。38.2°、44.3°、64.5°和 77.6°处分别对应的是 Au(PDF No.04⁃0784)的(111)、(200)、(220)和(311)晶面衍射峰[20],表明Au保持了原有的结构。结合SEM照片(图3)可见,单质 Au颗粒有效负载在g⁃C3N4基体上。3 341 cm−1处的宽红外吸收峰是N—H键的伸缩振动引起的(图6b)[13]。1 200~1 700 cm−1之间是 C—N键和碳氧六元杂环的伸缩振动吸收峰,吸收带在808 cm−1处为三嗪环的振动吸收峰[13]。图中并没有Au的吸收峰,这是因为Au颗粒对红外光谱没有响应。图6c为 g⁃C3N4和Au@g⁃C3N4支架的Raman谱图,类似于石墨,位于1 315 cm−1处的拉曼峰为D峰,472、713、752、982、1 154 和 1 226 cm−1处均为 g⁃C3N4中CN杂环的特征峰[21⁃22]。值得注意的是,由于Au的等离子共振效应引起了强烈的表面增强拉曼散射效应[23],Au@g⁃C3N4支架拉曼峰的强度显著高于 g⁃C3N4支架。

图6 g⁃C3N4和Au@g⁃C3N4支架的XRD图 (a)、FTIR谱图 (b)和Raman谱图 (c)Fig.6 XRD patterns(a),FTIR spectra(b)and Raman spectra(c)of g⁃C3N4and Au@g⁃C3N4scaffolds
图7为Au@g⁃C3N4支架的XPS谱图,Au@g⁃C3N4支架材料包含C、N、O和Au等4种元素,没有其它杂质。图中O1s峰的结合能为532.0 eV,对应于附着在Au@g⁃C3N4支架表面的水分子或表面羟基(—OH)[24]。C1s的高分辨谱图中的2个峰对应的结合能分别为284.70和288.35 eV。其中,284.70 eV处的峰归因于C—C键,而288.35 eV处的峰归属于sp2杂化碳形成的N—C=N键,N—C=N键是碳元素在g⁃C3N4中的主要存在形式[24]。图7c显示了N1s的高分辨谱图中的3个峰对应的结合能为398.30、399.80和401.20 eV,分别代表C—N=C、N—(C)3和N—H键[25⁃26]。图7d中结合能83.25和87.10 eV分别为Au@g⁃C3N4支架上 Au4f7/2和 Au4f5/2峰[8],Au4f7/2和 Au4f5/2的结合能差为3.85 eV,表明Au@g⁃C3N4支架上Au以Au0形式存在[20,26],负载在g⁃C3N4支架上的物质为单质Au。
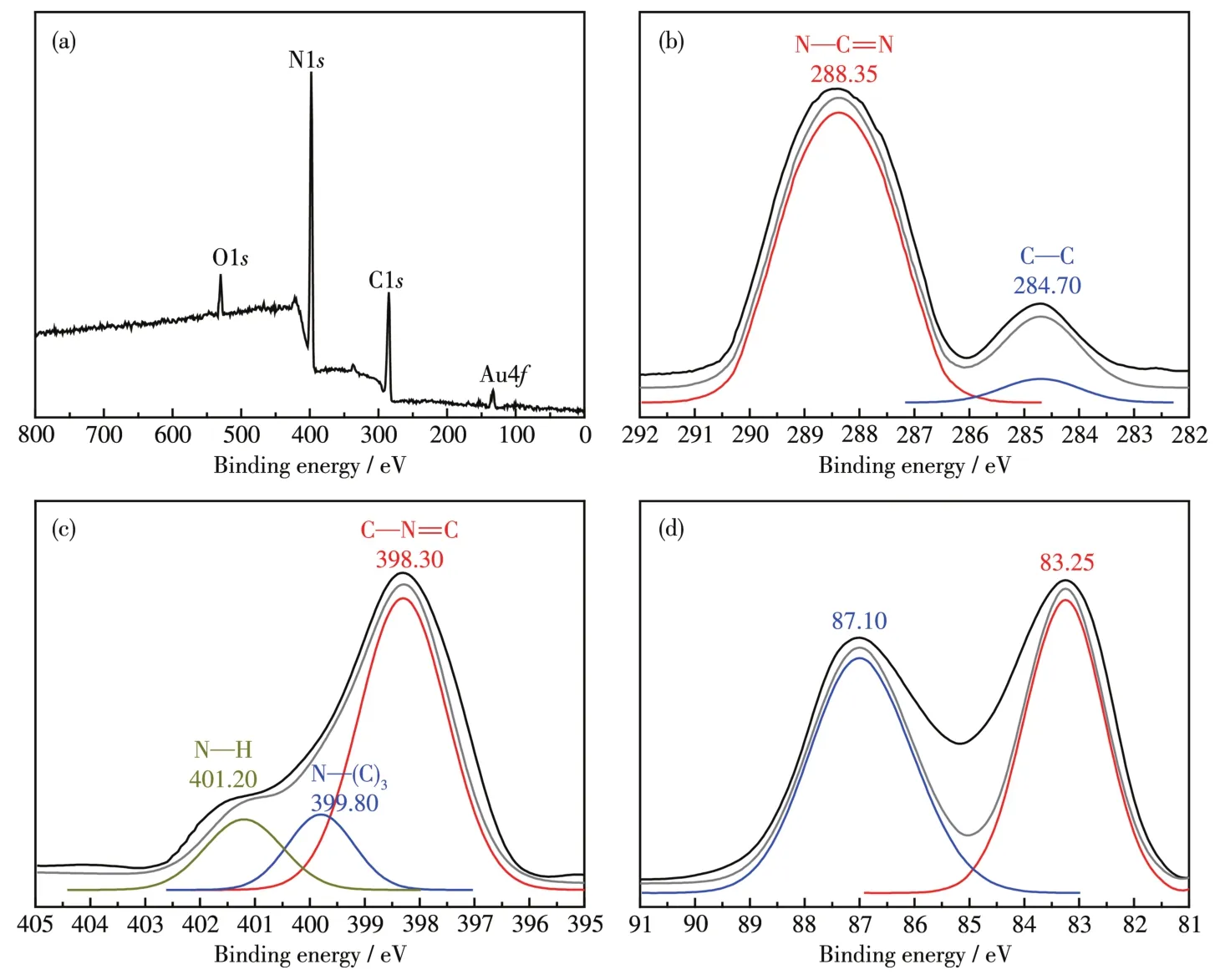
图7 Au@g⁃C3N4支架的XPS谱图Fig.7 XPS spectra of Au@g⁃C3N4scaffold
2.4 基本物性和力学性能
表1列出了MF和Au@g⁃C3N4支架的基本性能参数,MF支架的开孔率可达95%,比表面积达1 620 m2·g−1。SEM 照片表明热聚合后 Au@g⁃C3N4支架体积收缩(图2a和3a),孔径和纤维丝直径均显著降低(图 2b 和 3d),导致 Au@g⁃C3N4支架开孔率和比表面积分别降至 89% 和 1 480 m2·g−1(表 1)。但与块状 g⁃C3N4材料相比[2],Au@g⁃C3N4支架具有独特的三维网络状结构和更大的比表面积。

表1 MF和Au@g⁃C3N4支架基本性能参数Table 1 Basic property parameters of MF and Au@g⁃C3N4scaffolds
图8为MF、g⁃C3N4和Au@g⁃C3N4支架的应力−应变曲线,从图中可以看出,MF支架的拉伸强度达94.9 kPa,断裂形变量可达15.9%。热聚合后,g⁃C3N4和Au@g⁃C3N4支架的拉伸强度分别降至40.3和38.5 kPa,断裂形变量分别为9.6%和7.4%,Au@g⁃C3N4支架强度适中,依然具有一定韧性。
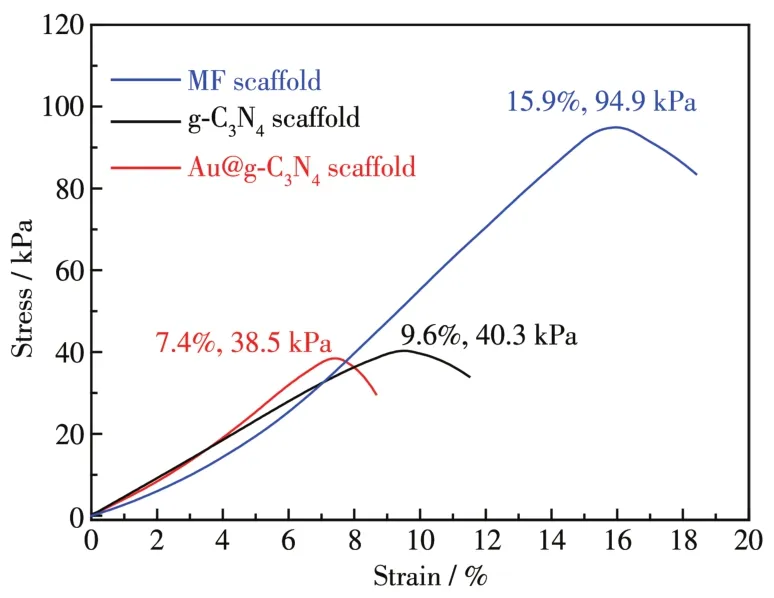
图8 MF、g⁃C3N4和Au@g⁃C3N4支架的应力−应变曲线Fig.8 Stress⁃strain curves of MF,g⁃C3N4,and Au@g⁃C3N4 scaffolds
2.5 光学性能分析
图9为g⁃C3N4和Au@g⁃C3N4支架的UV⁃Vis DRS谱图和PL谱图。g⁃C3N4和Au@g⁃C3N4支架在460 nm左右均出现了一个陡峭的吸收带边,这是由价带上的电子光激发跃迁至导带引起的,与热聚合三聚氰胺得到的 g⁃C3N4相似[2]。Au@g⁃C3N4支架在紫外可见光区域的光吸收强度相对较强,在550 nm处出现明显的吸收峰,主要归结于Au的表面等离子效应[16,18],这也导致Au@g⁃C3N4支架吸收带边发生红移[18,20]。g⁃C3N4和 Au@g⁃C3N4支架的吸收带边分别为 476 和507 nm,通过理论算法计算得到g⁃C3N4和Au@g⁃C3N4支架的带隙分别为2.61和2.45 eV[8]。在350 nm激发光下,由于 g⁃C3N4和 Au@g⁃C3N4支架受光激发后产生带边荧光现象[5,20],g⁃C3N4和 Au@g⁃C3N4支架在475 nm附近均出现了PL发射峰,但Au@g⁃C3N4支架的发射峰强度明显较弱,说明负载Au后,Au@g⁃C3N4支架中激发态电子−空穴对的复合率显著降低[18]。

图9 g⁃C3N4和Au@g⁃C3N4支架的UV⁃Vis DRS谱图(a)和PL光谱图(b)Fig.9 UV⁃Vis DRS spectra(a)and PL spectra(b)of g⁃C3N4and Au@g⁃C3N4scaffolds
2.5 光电化学分析
进一步研究了 g⁃C3N4和 Au@g⁃C3N4支架阻抗和瞬时电流特征(图10)。Au@g⁃C3N4支架的阻抗曲线半径更小,即阻抗较小(图10a),说明负载Au有利于降低传输电阻,Au@g⁃C3N4支架能更快速转移光生载流子,降低电子−空穴对复合[19]。在瞬时光照下,g⁃C3N4和 Au@g⁃C3N4支架均产生了一个激烈跳跃的光电流响应,g⁃C3N4和Au@g⁃C3N4支架的光电流分别为 0.28 和 0.62 μA·cm−2(图 10b)。与 g⁃C3N4支架相比,Au@g⁃C3N4支架能更快速地响应并产生更高的光电流,经过多次“开−关”光照反应,Au@g⁃C3N4支架光电流稳定,无明显衰减趋势。光电化学测试表明Au@g⁃C3N4将具有更加优异的光电响应特性。
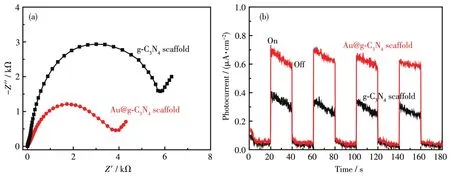
图10 g⁃C3N4和Au@g⁃C3N4支架的EIS谱图 (a)和瞬时电流曲线图 (b)Fig.10 EIS spectra(a)and transient photocurrent curves(b)of g⁃C3N4and Au@g⁃C3N4scaffolds
2.6 光催化性能
图11为g⁃C3N4和Au@g⁃C3N4支架对RhB的光催化降解和循环降解曲线图。在210 min内,空白组(没有添加催化剂)的RhB含量变化很小,说明RhB分子在可见光照射下非常稳定,几乎不发生自降解[10]。在暗反应过程中,开始的30 min吸附时间内,g⁃C3N4和 Au@g⁃C3N4支架表现出较强的吸附能力[16,27];最后30 min的吸附时间内,溶液中RhB浓度没有发生变化,说明其已达到吸附−脱附平衡,吸附量约为14%。在光照的条件下,g⁃C3N4支架对RhB具有催化降解能力,但降解率远低于Au@g⁃C3N4支架。210 min内g⁃C3N4和Au@g⁃C3N4支架对RhB的降解率分别为42%和82%。与g⁃C3N4支架相比,Au@g⁃C3N4支架催化降解率提高了近 1 倍(图 10)。从循环降解曲线(图11b)中可以看出,随循环次数的增加,光催化降解率虽有下降,但循环6次后光催化降解率降低幅度均未超过4%,表明g⁃C3N4和Au@g⁃C3N4支架均具有良好的可重复性和较高的化学稳定性[12]。由此可见,负载Au显著提高了Au@g⁃C3N4支架的光催化性能。

图11 g⁃C3N4和Au@g⁃C3N4支架对RhB的光催化降解 (a)和循环降解 (b)曲线图Fig.11 Photocatalytic degradation(a)and recycled degradation(b)profiles of RhB by g⁃C3N4and Au@g⁃C3N4scaffolds
在空气或惰性气体气氛下,通过500~600℃热聚合富氮前驱体可制备得到g⁃C3N4[4],过程虽然简单,但得到的均为块状g⁃C3N4[2],其比表面积较小(<20 m2·g−1)[28⁃29],且其光生电子−空穴对的复合率很高,使得g⁃C3N4量子产率和催化活性较低。众所周知,g⁃C3N4的比表面积越大,其表面活性位点越多,光催化活性也越高。采用模板法[18]、复合[6]、球磨[27]、降低维度[30]等手段可使g⁃C3N4比表面积增大至70~200 m2·g−1。本研究利用树脂前驱体的可塑性,采用发泡法构建三维网络状前驱体支架(图2),并负载了Au,最终得到负载Au的石墨相氮化碳(Au@g⁃C3N4)支架(图3、6和7),其比表面积增大至1 480 m2·g−1(表1)。Au@g⁃C3N4支架与反应物的接触面积更大,暴露更多的活性位点,提高g⁃C3N4的光催化性能。但也存在不足,如用三聚氰胺直接热聚合后一般得到淡黄色 g⁃C3N4,而 Au@g⁃C3N4支架呈灰色(图 3)。这是由于MF的碳氮比率过高,导致Au@g⁃C3N4支架中可能存在一定的无定型碳。后续还有待引入更高氮含量的前驱体,调控MF的碳氮比率,以期得到更纯的g⁃C3N4。
光催化降解率关键在于光生载流子的分离效率,而g⁃C3N4由C和N原子相互交替构成,导电性不佳,光生载流子迁移需要巨大的能量,光生电子−空穴对的快速复合是g⁃C3N4光催化性能不佳的根本原因[3,29]。抑制g⁃C3N4电子和空穴的复合是提高其光催化性能的根本途径。Au和g⁃C3N4具有不同的费米能级,g⁃C3N4的费米能级较高,而Au的费米能级较低,当Au与g⁃C3N4之间产生有效的表面接触后,光生电子会从g⁃C3N4迁移到Au上,形成捕获光生电子的肖特基势垒[31],从而加大载流子分离效率,有效抑制g⁃C3N4光生电子−空穴对的复合(图7b)。我们成功地将Au粒子均匀负载在g⁃C3N4支架上,构建了Au@g⁃C3N4支架(图 1 和 3),实现了 Au和 g⁃C3N4有效复合(图5和6);此外,由于MF支架开孔率高(图2和表1),溅射的Au能穿过孔道,可到达支架内部(图2e);调节溅射时间可以调控Au的负载量,通过多次实验表明,溅射90 s为最佳工艺参数,Au的负载量达 6%(图 5),此时Au@g⁃C3N4支架具有最优性能。通过 PL 谱图(图 9b)、EIS 谱图(图 10a)和瞬时电流曲线图(图10b)均证明了Au的负载有效抑制了Au@g⁃C3N4支架光生电子−空穴对的复合[32]。
自然光中紫外光(<380 nm)仅占比约4%,而自然光中主要能量集中在可见光,可见光(380~780 nm)约占自然光总能量的45%,然而g⁃C3N4的可见光吸收和利用率较低[4]。Au具有表面等离子体共振效应(图6和9),对可见光具有较强的吸收能力[11],负载Au拓宽了Au@g⁃C3N4支架的光吸收范围,在550 nm出现了新吸收峰,Au@g⁃C3N4支架的吸收带边从460 nm红移至507 nm,禁带宽度缩小至2.45 eV。因此,负载Au既抑制了电子−空穴对复合,又提高了光吸收,显著提高了Au@g⁃C3N4支架光催化性能,与g⁃C3N4支架相比,Au@g⁃C3N4支架的光催化降解RhB效率提高近1倍(图11)。g⁃C3N4粉体与水中的污染物接触面积大、效率高,但粒径小,难于从水体中分离、回收和循环利用。Au@g⁃C3N4支架在保证了与水中污染物较大的接触面积的情况下,自身具有一定的强度和韧性(图8),不易破损,便于从水体中分离、回收和循环利用,催化稳定性强(图11),也适合做成各种光催化器件。
3 结 论
以三聚氰胺等原料合成具有可塑性的MF,通过发泡法构建前驱体支架,并通过磁控溅射法沉积Au,550℃下热聚合后,成功制备得到负载Au的石墨相氮化碳(Au@g⁃C3N4)支架,其比表面积可达1 480 m2·g−1。负载 6%Au 后,Au@g⁃C3N4支架在 550 nm处出现了新的吸收峰,吸收带边红移至507 nm,禁带宽度为2.45 eV。同时,Au@g⁃C3N4支架荧光强度和阻抗显著降低,光电流从 0.28 μA·cm−2提升至0.62 μA·cm−2。金的负载既拓宽了 Au@g⁃C3N4支架可见光吸收,又抑制了电子−空穴对的复合,光催化性能稳定,光催化降解率提高近1倍。此外,Au@g⁃C3N4支架拉伸强度达38.5 kPa,拉升率达7.4%,不易破碎,在光催化降解水体中有机污染物应用中便于回收和再利用。
