NiMoP/C复合材料的制备及其电催化析氢性能
2022-04-15杨占旭
宋 楗,韩 乔,杨占旭
(辽宁石油化工大学石油化工学院,辽宁 抚顺 113001)
在经济全球化的整体趋势下,人类社会进入了能源消费时代[1⁃3]。现代生活和工业离不开能源,然而仅仅依靠传统的不可再生化石能源,远不能满足当前现代工业进一步发展的需求,此外燃烧化石燃料会带来严重的环境问题,例如酸雨、温室气体排放等[4⁃5]。因此,以氢能为代表的可再生清洁能源的开发利用受到了研究者的高度重视。
氢能是最有可能代替化石能源的后备资源,能够满足当前对可持续能源系统的需求。传统的制氢方式多为工业蒸汽重整,该方法的缺点为能耗高、转化效率低,并且会排放CO2,每生产1 kg的H2就会有13.7 kg CO2被排放到大气中[6]。相比之下,电解水制氢方式因其绿色环保且能够持续高效产氢的优势逐渐受到研究者的关注[7]。Pt基等贵金属催化剂虽然具备好的电催化析氢(Hydrogen Evolution Reaction,HER)催化活性,但储量稀少、成本昂贵,这限制了其在工业上的开发应用[8⁃10]。近年来,研究者一直致力于寻找和制备各种高效非贵金属催化剂,其中以Mo基廉价过渡金属为核心的化合物作为析氢反应的催化材料表现出较强的优势。目前已经广泛研究了许多Mo基化合物,包括Mo基合金(Ni⁃Mo、Fe⁃Mo、Co⁃Mo等)[11]、MoS2[12]、MoP[13]、MoC[14]、MoN[15]、MoO2[16]、MoB[17]等。M.M.Jaksic等[18]发现Ni和Mo之间存在低⁃超⁃d轨道电子的相互作用,从而产生协同效应来增强HER活性。H.E.Ebtesam等[19]首次以胶体形式合成了双金属NiMoP纳米颗粒,通过控制Ni/Mo物质的量比来制备不同的晶体结构,并考察了其碱性条件下的HER性能。结果表明,NiMoP材料在碱性溶液中表现出优异的HER析氢性能和很高的稳定性。通过构建特殊纳米结构的NiMoP,将其与碳等导电材料复合使用,能够增加催化剂的比表面积,使催化剂表面暴露出更多的活性位点,进而改善NiMoP颗粒的分散性,以实现更加优越的性能。
据研究报道[20⁃21],NiMo合金是在碱性条件下HER活性最高的电催化剂,由于其耐腐蚀性差,在酸性介质中的研究很少。基于此,本研究采用溶液凝胶法合成催化剂的前驱体,对前驱体在基底上的分散性进行调控,再通过化学气相沉积法在N2保护条件下对前驱体进一步磷化得到了NiMoP/C催化剂。样品纯相MoP/C与Ni2P/C材料的制备步骤也与以上报道相似,不同之处在于制备过程中不加Ni源或Mo源。同时采用电化学测试方法研究了以上材料在酸性介质中的电催化析氢活性。
1 实验部分
1.1 试剂
四 水 合 钼 酸 铵((NH4)6Mo7O24⋅4H2O)、六 水 合 硝酸镍(Ni(NO3)2⋅6H2O)、萘酚Nafion溶液(C10H8O),分析纯,上海阿拉丁试剂公司;磷酸(H3PO4),优级纯,沈阳市东兴试剂厂;三乙醇胺(C6H15NO3),优级纯,天津市恒兴化学试剂制造有限公司;一水柠檬酸(C6H8O7⋅H2O,优级纯)、蔗糖(C12H22O11,分析纯),国药集团化学试剂有限公司;无水乙醇(C2H5OH),分析纯,天津市大茂化学试剂厂;硫酸(0.5 mol/L),自制;盐酸(1 mol/L),自制;高纯N2,抚顺气体厂;去离子水,自制。本实验所用药品均未经纯化处理。
1.2 仪器
实验仪器:BSA 124S⁃CW电子天平,德国赛多利斯科学仪器有限公司;DF⁃101S恒温加热磁力搅拌器,巩义市予华仪器有限公司;XMTD⁃400电热恒温水浴锅,北京市永光明医疗仪器公司;GZX⁃9070MBE数显鼔风干燥箱,上海博讯实业有限公司医疗设备厂;GSL⁃1400X双温区真空管式炉,合肥科晶材料技术有限公司。
表征仪器:XRD,D8 Advance X射线衍射仪,德国Bruker公司,测试条件为Cu靶⁃Kα射线,入射波长λ=0.154 nm,管电压40 kV,管电流40 mA,扫描速 率2θ=5(o)/min,扫 描 范 围2o~70o;XPS,ESCALAB 250 X射线光电子能谱仪,美国赛默飞世尔科技公司;SEM,Hitachi SU 8010,日本日立公司;TEM,JEM 2100F,日本电子株式会社;DXR,拉曼分析仪,美国赛默飞世尔科技公司;BET,Autosorb⁃IQ2⁃MP全自动比表面积和孔径分析仪,美国康塔仪器有限公司;VSP⁃300多通道电化学工作站,法国Biologic公司。
1.3 催化剂的制备
先采用溶液凝胶法合成前驱体,再用程序升温煅烧得到最终产物,具体步骤如下:
(1)称量0.177 g四水合钼酸铵、0.29 g六水合硝酸镍、0.59 g三乙醇胺、0.12 g磷酸、0.21 g一水柠檬酸、0.34 g蔗糖溶于10 mL去离子水中,室温下磁力搅拌10 min至溶液混合均匀,得前驱体溶液。
(2)将前驱体溶液置于90℃水浴中加热处理4 h以上,直至溶剂水挥发完全。在真空干燥箱中120℃烘干过夜,得前驱体干凝胶。将所得干凝胶放入刚玉舟中,于管式炉中煅烧,在N2保护气氛下,以5℃/min的速率升温到800℃并恒温焙烧2 h,待炉管自然冷却后,取出样品,研磨备用。所得粉末即为NiMoP/C材料。
对比样品的制备步骤同上,不同之处在于纯相MoP/C材料制备过程中未加入Ni源,纯相Ni2P/C材料制备时未加入Mo源。
1.4 工作电极的制备
将碳纸整齐裁剪为1.0 cm×1.5 cm的长方形,并放置在装有10 mL 1 mol/L的HCl溶液的50 mL烧杯中,超声洗涤10 min,以去除表面可能附着的杂质。再换用无水乙醇溶液继续超声洗涤10 min,超声结束后将其固定在电极夹上,并用红外灯烘干碳纸表面,备用。
将10 mg合成的催化剂粉末均匀分散在含有450μL无水乙醇和50μL萘酚溶液(体积比9∶1)的混合溶液中,超声30 min,使用移液枪将50μL的分散液滴加在碳纤维纸上,按照相同的方法共移取6次,红外灯下烘烤挥发乙醇去除溶剂,得到表面均匀覆盖活性物质的工作电极,负载量是6 mg/cm2。
所有电化学测试均使用VSP⁃300电化学工作站在标准三电极体系下进行,以饱和甘汞电极(SCE)为参比电极,石墨棒为对电极,预先配制好0.5 mol/L H2SO4溶液,移取50 mL加到标准三电极反应器内。所有测试均未经过IR校正,实验中所测得的过电位均以标准氢电极校准,表示为:
E(RHE)=E(SCE)+0.059p H+0.244(V)(1)
2 结果与讨论
2.1 材料形貌和结构表征
2.1.1 XRD表征 图1是NiMoP/C、对比样品MoP/C和Ni2P/C的XRD衍射图谱。由图1可知,在39.2o、43.2o、47.3o、54.2o的 衍 射 峰 依 次 对 应Ni⁃MoP的(111)、(201)、(210)、(300)晶面,与NiMoP的标准卡片PDF#31⁃0873相吻合[21];在32.2o、43.1o、67.0o处的衍射峰分别对应MoP的(100)、(101)、(200)晶面,对应MoP的标准卡片PDF#24⁃0071[22];在40.8o、44.6o、47.3o处的衍射峰依次对应Ni2P的(111)、(201)、(210)晶面,对应Ni2P的标准卡片PDF#03⁃0953[22]。在图1中没有观察到其他杂峰,说明成功合成了纯相NiMoP/C、Ni2P/C、MoP/C复合材料。

图1 NiMoP/C、MoP/C、Ni2P/C材料的XRD图谱Fig.1 XRD patterns of NiMoP/C,MoP/C和Ni2P/C
2.1.2 SEM、TEM和HRTEM表征 图2给出了不同材料的SEM、TEM和HRTEM图像。

图2 Ni2P/C、MoP/C、NiMoP/C的SEM、TEM和HRTEMFig.2 SEM,TEM and HRTEM images of Ni2P/C,MoP/C and NiMoP/C
由图2(a-f)可知,Ni2P/C、MoP/C、NiMoP/C材料的形状均为块状,材料的主体是碳基底,合成的材料随机分布在基底上,但尺度均小于1μm,属于纳米颗粒材料,该结构能提供更多的边缘暴露位置,增加活性位点的数量,易于与电解质溶液接触,因而能提高材料的电化学析氢催化性能。图2(jl)分别对应图2(g-i)中绿色区域的HRTEM图像。其中,图2(j)中晶面间距为0.22、0.50 nm的晶格条纹对应Ni2P的(111)和(100)晶面[22];图2(k)中MoP/C材料中只有一种晶面间距为0.32 nm的晶格条纹对 应MoP的(001)晶 面[22];图2(l)中 晶 面 晶 距 为0.22、0.24 nm的晶格条纹对应NiMoP的(111)和(201)晶面[21]。以上表征结果说明成功制备出了Ni⁃MoP/C、Ni2P/C、MoP/C复合材料。
图3为不同材料的EDS能谱分析。从图3可知,在Ni2P/C、MoP/C和NiMoP/C的EDS元素分布中,Mo、Ni、P各元素都均匀附着在C基底表面。证明制备的材料纯度较高,无其他杂元素的干扰。

图3 Ni2P/C、MoP/C、NiMoP/C的EDS能谱Fig.3 EDSenergy spectrum of Ni2P/C,MoP/C,NiMoP/C
2.1.3 XPS表征 通过XPS测试进一步研究了NiMoP/C复合材料表面的元素组成,结果如图4所示。由图4(a)XPS总谱可知,Ni2P/C主要由Ni、P和C三种元素组成,MoP/C主要由Mo、P和C三种元素组成,NiMoP主要由Ni、Mo、P和C四种元素组成,与EDS能谱分析结果一致,在XPS谱图中未见其他元素,进一步证明合成的Ni2P/C、MoP/C和NiMoP/C材料纯度较高。
由图4(b)可以看出,C 1s的XPS光谱可分为三组不同衍射峰,首先是Ni2P、MoP和NiMoP在284.80 eV处的衍射峰均对应于C=C,归因于以sp2方式存在的石墨化碳。其次,Ni2P在286.51 eV处的衍射峰、MoP在286.28 eV处的衍射峰、NiMoP在286.14 eV处的衍射峰均归属于离子型的C-O,最后Ni2P在288.25 eV处的衍射峰、MoP在289.08 eV处的衍射峰、NiMoP在288.84 eV处的衍射峰均对应于C=O。
由图4(c)可以看出,Ni2P在531.41 eV处的衍射峰、MoP在531.66 eV处的衍射峰、NiMoP在531.4 eV处的衍射峰均归属于O-P,主要以PO2-4的形式存在,这是因为在空气中各材料表面的P被氧化形成了磷酸盐[21];Ni2P在533.40 eV处的衍射峰、MoP在533.34 eV处的衍射峰、NiMoP在533.04 eV处的衍射峰则可归因于被吸附的游离态的O物种。强O信号的出现是因为材料表面存在的吸附氧或是由于暴露在空气中的过渡金属磷化物的氧化或是磷酸盐的氧化造成的。
由图4(d)可以看出,在Ni2P/C中134.86 eV和133.06 eV(P5+2p1/2、2p3/2)处的两个衍射峰、MoP/C中134.01 eV(P5+2p3/2)处的衍射峰、NiMoP/C中133.79 eV(P5+2p3/2)处的衍射峰,应该都归属于氧化态的PO3-4或P2O5,可能是材料表面氧化形成的磷氧化物;在Ni2P/C中130.44 eV(Pδ-2p1/2)处衍射峰归属于Ni-P[22],MoP/C中130.59 eV和129.67 eV(Pδ-2p1/2、Pδ-2p3/2)处 的 两 个 峰 归 属 于Mo-P[22],NiMoP/C中130.16 eV(Pδ-2p1/2)处的衍射峰归属于直接与Mo原子作用的低价态的P[21]。P的引入可以降低金属原子自身的电子云密度,减弱金属氢键之间键能,从而利于吸附态的Hads从金属表面的活性位点脱附形成氢气[22]。
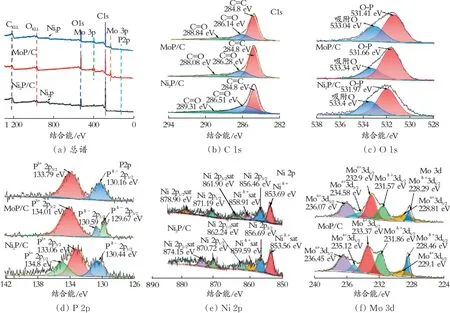
图4 催化剂的XPS谱图Fig.4 Fitted XPSspectra for as⁃prepared catalysts
由图4(e)可以看出,在Ni2P中853.56 eV和859.59 eV(Niδ+2p3/2、Niδ+2p3/2sat)处 的两个衍 射 峰、NiMoP中853.69 eV和858.91 eV(Niδ+2p3/2、Niδ+2p3/2sat)处的两个衍射峰都对应于Niδ+(0<δ≤2),归属于Ni-P;Ni2P中856.69 eV和870.72 eV(Ni 2p3/2、Ni 2p1/2)处的两个衍射峰及其对应的862.24 eV和874.15 eV(Ni 2p3/2sat、Ni 2p1/2sat)处的宽振荡峰和NiMoP中位于856.46 eV和871.19 eV(Ni 2p3/2、Ni 2p1/2)处的两个衍射峰及其对应的861.90 eV和878.90 eV(Ni 2p3/2sat、Ni 2p1/2sat)处 的振荡峰,根据文献[21]报道,可以归属于以氧化态的NiO、Ni(OH)2、NiOOH和磷化物等形式存在的Ni-O或Ni-P。
由 图4(f)可 以 看 出,MoP在228.46 eV和231.86 eV处、NiMoP在228.29 eV和231.57 eV处的特征峰,都归属于MoP或NiMoP中处于低价态的Moδ+(0<δ≤2);MoP在236.45 eV和233.37 eV处、NiMoP在236.07 eV和232.90 eV处的特征峰,对应于高氧化态的MoO3中的Mo6+,MoP在229.10 eV和235.12 eV处的特征峰、NiMoP在228.81 eV和234.58 eV处的特征峰,对应于MoO2中的Mo4+。
从图4(e)中的Ni2p高分辨XPS图谱中可以看出,与纯Ni2P中Ni 2p(853.56 eV)的结合能相比,NiMoP/C中Ni 2p的结合能(853.69 eV)向高结合能方向移动了0.13 eV,这表明电子从Ni移动到了Mo,这是由于Ni和Mo周围的电子云密度增加,电负性增强,导致电子重新排布。从图4(f)中的Mo 3d高分辨XPS图谱中可以得出,与MoP/C中Mo 3d(228.46 eV)的结合能相比,NiMoP/C中Mo 3d的结合能(228.29 eV)向低结合能方向移动了0.17 eV,这一结果表明Mo原子的电负性增加,部分电子从Ni转移到Mo,与Ni 2p的结合能移动互为印证。
综合可以推测,电子在NiMoP/C纳米复合材料之间的转移要比在MoP/C和Ni2P/C材料中更为容易,同时Ni⁃Mo之间具有强烈的相互作用,这对于提高材料的HER性能具有重要意义。
2.1.4 Raman表征 图5为Ni2P/C、MoP/C和NiMoP/C复合材料的Raman谱图。

图5 催化剂的Raman谱图Fig.5 Raman spectra for as⁃prepared catalysts
如图5所示,D⁃峰代表C原子晶格的缺陷,G⁃峰代表C原子sp2杂化的面内伸缩振动模式,其强度的比值多用来分析催化剂中C的石墨化程度。经过分峰拟合和计算得出,纯Ni2P/C、MoP/C和NiMoP/C复合材料的ID/IG分别是3.558、3.531和3.075,这表明三者中的碳多为无定形碳结构,与Ni2P/C和MoP/C材料相比,NiMoP/C材料的石墨化程度相对较高,由此可知NiMoP/C复合材料的导电性要好于纯Ni2P/C和MoP/C材料。
2.1.5 N2吸附⁃脱附等温线 图6(a)为Ni2P/C、MoP/C和NiMoP/C的N2吸附⁃脱附等温线。由图6(a)可知,各材料的吸附⁃脱附等温线都具有IV型等温线特征,说明样品具有介孔性质。NiMoP/C、Ni2P/C和MoP/C材料的比表面积分别为299、259、106 m2/g。由图6(b)的孔径分布可知,3种复合材料的孔径较小,主要集中分布在1~5 nm。综合以上分析,NiMoP/C复合材料具有更大的比表面积与介孔结构,大的比表面积有利于增大电解液与工作电极之间的接触面积,能够提供更多活性位点,有利于电子传输和提升材料的电解水析氢性能。

图6 催化剂的N 2吸附⁃脱附等温线和孔径分布Fig.6 Nitrogen adsorption⁃desorption isotherms and pore size distribution for as⁃prepared catalysts
2.2 线性扫描(LSV)极化曲线测试
图7(a)为在0.5 mol/L H2SO4电解液中测试不同材料的线性扫描(LSV)极化曲线。当电流强度为-10 mA/cm2时,NiMoP/C、Ni2P/C、MoP/C催化剂的过电势分别为-158、-192、-228 mV(vs RHE)。图7(b)为由LSV曲线拟合得到的Tafel斜率曲线。由图7(b)可知,NiMoP/C电催化剂具有较好的HER催化活性。NiMoP/C的Tafel斜率(111 mV/dec)均 比Ni2P/C(116 mV/dec)、MoP/C(174 mV/dec)的Tafel斜率小,说明NiMoP/C材料具有更好的导电性能。图7(c)为由Tafel斜率推算的起始交换电流密度,NiMoP/C材料的起始交换电流密度为0.251 1 mA/cm2,比MoP/C的起始交换电流密度0.487 8 mA/cm2小很多,但比Ni2P/C的起始交换电流密度(0.221 2 mA/cm2)要高,这说明在HER反应的初始阶段,纯相Ni2P/C材料更容易释放出H2,然而随着起始交换电流密度的增大,NiMoP/C材料逐渐显出优越的电催化析氢活性。图7(d)为各材料在10 mA/cm2电流密度时的析氢过电位η10和起始交换电流密度j0的数据对比柱状图,可以看出NiMoP/C材料的电催化析氢性能比Ni2P/C和MoP/C材料更为优异。

图7 催化剂电催化HER的相关数据对比Fig.7 Data and graphs related to as-prepared catalysts for electrocatalytic HER
2.3 电化学阻抗谱(EIS)测试测试
为了进一步了解NiMoP/C催化剂的电荷转移情况,对其进行了电化学阻抗谱(EIS)测试,结果如图8所示。图8中半圆的半径越小,则电荷转移电阻越小,经过拟合计算得到NiMoP/C、Ni2P/C和MoP/C复合材料的电荷转移电阻分别为4.968、5.788、8.025Ω。该结果表明,NiMoP/C复合材料在电催化HER过程中有着更快的电荷传输速率,从而表现出更优异的HER催化性能。

图8 催化剂的电化学阻抗谱Fig.8 Nyquist images for as⁃prepared catalysts
电化学活性面积(ECSA)是HER反应的一个非常重要的影响因素,因为ECSA的增加往往导致催化活性的提高。为了进一步说明NiMoP/C材料的催化活性,采用循环伏安法(CV)对各催化剂的双电层电容(Cdl)进行了测量,结果如图9所示。

图9 催化剂在不同扫描速率下的双电层电容Fig.9 C dl of different scan rates for as prepared catalysis
由图9可见,ECSA与Cdl具有线性关系。NiMoP/C的Cdl为45.04 mF/cm2,较Ni2P/C和MoP/C的Cdl(31.44、21.06 mF/cm2)都 大,假 设 以MoP/C的Cdl所对应的ECSA为1.000 cm2,则NiMoP/C和Ni2P/C复合材料所对应的ECSA分别为2.138 cm2和1.495 cm2,说明NiMoP/C材料的催化性能更好。
2.4 稳定性测试
对NiMoP/C、Ni2P/C、MoP/C材料的稳定性进行了测试,结果如图10(a)所示。经过1 000圈的循环之后,NiMoP/C的极化曲线较之前要低,原因可能是在1 000圈循环后NiMoP/C材料被电化学活化,析氢性能有所提升。纯Ni2P/C循环前后曲线几乎重合,而MoP/C循环后的性能比循环前要差很多。图10(b)为经过40 000 s恒电流密度(100 mA/cm2)测试的计时电位曲线。由图10(b)可见,NiMoP/C的析氢过电位比刚开始时降低了28 mV,Ni2P/C降低了19 mV,而MoP/C增加了21 mV,析氢过电位的降低是HER性能优化的过程,说明在电化学活化过程中NiMoP/C相比于Ni2P/C、MoP/C的电催化HER性能和稳定性得到提升[23⁃25]。

图10 催化剂在1 000圈扫描前后的极化曲线和计时电位测试曲线Fig.10 LSV curves for as⁃prepared catalysts before and after 1 000 cycles and chronopotential test curves
3 结 论
采用简易有效的方法制备了块状NiMoP/C复合材料,研究表明,相比纯相的Ni2P/C和MoP/C材料,NiMoP/C材料的电化学析氢活性最高,其在电流密度为-10 mA/cm2的过电位为-158 mV,Tafel斜率为111 mV/dec,并且有着良好的稳定性。与纯相Ni2P/C和MoP/C相比,NiMoP/C材料催化性能优异的原因是:NiMoP纳米颗粒具有优越的本征HER活性,碳材料具备良好的导电性,此外材料本身具有的大比表面积和丰富的介孔结构,能暴露出更多的活性位点,从而有效增强NiMoP/C复合材料的电化学析氢催化活性。同时电化学活化过程增强了Ni和Mo金属原子之间的电子协同效应,可以提升反应过程中的电荷转移速率,进一步提高了NiMoP/C催化剂的电解水HER催化活性和稳定性。
本研究利用双过渡金属原子掺杂的方式制备NiMoP/C复合材料,研究了催化剂在酸性条件下的HER电催化活性和工作稳定性,为未来开发高效稳定的非贵金属析氢催化剂提供了一种新的思路。
