替考拉宁和万古霉素高效液相色谱手性固定相的研究
2022-04-05刘金月祝金艳袁黎明
刘金月,苏 鹏,祝金艳,艾 萍,袁黎明
(云南师范大学 化学化工学院,云南 昆明 650500)
所谓手性就是像人的左手与右手,互为实物与镜像的关系,两两不能完全重合。如同手的这一特征,也广泛存在于其他事物中,并且在生命的产生和演变过程中也有所体现[1]。分子是否是手性分子,其本质是要判断分子有无对称面和对称因素[1]。据统计,目前用于临床的药物种类约有3 500~4 000种,其中很大一部分药物,是通过合成或半合成的生产方式制备的,在这些药物中,约有40%以上并非仅由一种对映体组成,通常都是外消旋体[2]。在外消旋体药物中,在大多数情况下只有其中的一种立体异构体可用于临床治疗,而它的另一种旋光异构体可能根本没有疗效,或者具有完全相反的药效,甚至可能具有一定的毒副作用,对机体造成一些不可逆的损伤。随着生活水平的提高,人们对低毒性高效用的药品需求也越来越大,同时为了避免类似“反应停”悲剧事件的重演,各国特别是美国、日本、法国等国都极其重视对外消旋药物的拆分[3-8]。采用现代分析方法进行临床医学样本的分析检验,已经成为生命科学中的一个重要领域,具有极大的临床医学价值[9]。早在20世纪90年代初,美国相关部门就规定:只要是研究具有旋光异构体的药物,必须对它的每一个对映体进行药理和毒理性质研究和评估[10]。研究如何获得纯的对映体、并研究单一手性化合物的性质具有重要的实用价值[11-13]。制备单一手性的手性固定相拆分外消旋体[14]与已有的手性分离的方法相比,HPLC因其高效性、低成本和重现性而被认一种合适的方法,也被广泛应用在生产生活的各个领域[15-18]。
替考拉宁和万古霉素都是糖肽类大环抗生素,它们的结构上含有羟基、羧基、氨基等多种官能团及孔穴结构,并具有良好的立体选择性,是非常有效的手性识别材料[19-20]。用不同的键合臂分别制备了四种HPLC替考拉宁和万古霉素CSP,并在反相模式和极性有机相模式下考察了这八种手性柱的性能。结果表明,固定相制备方法的不同和色谱条件的改变对色谱柱拆分性能有一定的影响。不相同的键合臂,能拆分的对映异构体可能不一样,这些柱之间具有一定的互补性,自制的八种色谱柱在手性分离方面有一定的可行性。
1 实验部分
1.1 仪器与试剂
仪器:DF-101S 集热式恒温加热磁力搅拌器(巩义市予华仪器有限责任公司);Model 1666 Slurry Packer 高效液相色谱装柱机(美国Alltech公司);高效液相色谱仪(大连依利特);XL30ESEM-TMP型扫描电子显微镜(荷兰飞利浦公司);PHS-25雷磁 pH计(上海精密科学仪器有限公司);高纯水机(英国ELGA Lab Water);AL204 梅特勒-利托多电子天秤(上海梅特勒-利托多仪器有限公司);DHG-9035A 电热鼓风干燥箱(上海一恒科学仪器有限公司);EYELA 磁力搅拌低温恒温水槽(PSL-1810,上海爱朗仪器有限责任公司);紫外-可见检测器(SPD-15C,日本岛津);高压恒流输液泵(LC-15C,日本岛津);液相色谱匀浆装柱机(液相色谱匀浆装柱机,美国Alltech公司);台式低速离心机(TDZ5-WS,湘仪公司);超声波清洗仪(As3120,天津奥特赛恩斯仪器有限公司)。
试剂:色谱硅胶(Unisil 5-120,苏州纳微科技有限公司);替考拉宁(苏州法母进出口有限公司);3-(2,3-环氧丙氧)丙基三甲氧基硅烷(>99%,Adamas试剂公司);3-巯基丙基三甲氧基硅烷(>99%,Adamas试剂公司);偶氮二异丁腈(AIBN)(>99%,Adamas试剂公司);10-十一碳烯酰氯(>99%,Adamas试剂公司);1,6-己二异氰酸酯、异氰酸丙基三乙氧基硅烷(>99%,上海阿达玛斯试剂公司);4, 4′-二苯基甲烷二异氰酸、3-氨丙基三乙氧基硅烷(>99%,上海西格玛奥德里奇贸易有限公司);YQG80型球形硅胶(粒径5 μm,购买于中国青岛美高化工有限公司);万古霉素在苏州法母进口有限公司购买;2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉(EEDQ,99%,上海共价化学科技有限公司);手性化合物均购自东京化成工业株式会社;其余试剂均为分析纯,均购于天津市风船化学试剂科技有限公司。
1.2 固定相制备
替考拉宁手性固定相CSP-1的制备:参考文献[21]的方法,称取1.6 g替考拉宁,用25 mL 无水吡啶-无水DMSO (1∶1,体积比,下同),于60 ℃搅拌直至替考拉宁完全溶解后,加入2 g已经活化好的硅胶,用超声处理5 min 后,在N2保护下,缓慢加入0.4 mL异氰酸丙基三乙氧基硅烷,100 ℃反应12 h,冷却后分别用DMF、乙腈、甲醇洗涤反应物,离心洗涤数次,60 ℃真空干燥至恒重备用。
替考拉宁手性固定相CSP-2的制备:参考文献[22]的方法,同CSP-1的方法将替考拉宁溶解,在室温下,小心加入0.3 mL 10-十一碳烯酰氯25 ℃反应24 h,充分反应后,向上述冷却至室温的溶液中,加入2.0 g已经巯基化的硅胶和0.1 g偶氮二异丁腈,在N2保护下,升高温度酯60 ℃,并保持24 h,待反应体系冷却至室温后,洗涤、干燥方式同上,产物备用。
替考拉宁手性固定相CSP-3的制备:参考文献[22]的方法,同CSP-1的方法将替考拉宁溶解,在室温下,小心加入0.2 mL甲基丙烯酸异氰酸乙酯60 ℃反应24 h,反应结束冷却至室温,在上述溶液中加入2 g巯基化硅胶和0.1 g偶氮二异丁腈,氮气保护下60 ℃反应24 h,洗涤、干燥方式同上,产物备用。
替考拉宁手性固定相CSP-4的制备:参考文献[23]的方法,称取1.6 g替考拉宁,用无水DMF溶解,加入已经环氧丙氧基化的硅胶2 g,100 ℃反应12 h,反应结束冷却至室温后,洗涤、干燥方式同上,产物备用。
万古霉素手性固定相CSP-A的制备:将已经氨丙基化的3 g硅胶中放入圆底烧瓶中,在冰浴、N2保护下加入50 mL干燥过的甲苯、2.5 mL1,6-己二异氰酸酯,将混合物在70 ℃油浴中反应2 h,关掉加热开关,将装置从油浴中提起,产物冷却至25 ℃,产物分别用吡啶、乙腈、二氯甲烷洗涤多次,在60 ℃下减压干燥备用。
万古霉素手性固定相CSP-B的制备:在N2保护下,依次在圆底烧瓶中加入2.5 g氨丙基硅胶、10 mL无水DMF、4, 4′-二苯基甲烷二异氰酸酯。将混合物在100 ℃油浴中反应2 h后,待产物冷却至25 ℃,用DMF离心洗涤3遍后移入烧瓶中,在N2保护下,加入万古霉素和25 mL无水DMF,在100 ℃下反应24 h,反应结束后丙酮、乙醇、甲醇洗涤多次,产物在60 ℃下真空干燥备用。
万古霉素手性固定相CSP-C的制备:在N2保护下,依次在圆底烧瓶中加入4 g氨丙基硅胶、万古霉素、EEDQ和20 mL无水DMF。将混合物在100 ℃油浴中反应6 h后,待产物冷却至25 ℃,依次用水、甲醇洗涤干净,将固体于60 ℃ 抽真空干燥过夜,产物备用。
万古霉素手性固定相CSP-D的制备:用50 mL无水吡啶-无水DMSO (1∶1)溶解万古霉素成悬浮液,取2 g酸活化过的硅胶在圆底烧瓶中,在冰浴、N2保护下,加入溶解好的万古霉素悬浮液和少量的异氰酸丙基三乙氧基硅烷,将混合液在100 ℃油浴中反应24 h,待产物冷却至25 ℃,分别用甲醇、乙腈、二氯甲烷洗涤,最后在60 ℃下干燥,产物备用。
1.3 高效液相色谱柱的制备
采用高压匀浆法,将上述制备好的八种固定相过250目筛,分别称取1.2 g固定相,以甲醇-水(90∶10,体积比,下同)溶液为顶替液,在压力为40~50 MPa下装柱,制得2.0 mm×250 mm的色谱柱。
1.4 仪器工作条件
检测波长210~254 nm,流动相为甲醇-水(80∶20)、甲醇-水(60∶40)、甲醇-三乙胺(1 000∶0.03)、甲醇-冰醋酸(1 000∶0.03),溶液流速为0.1 mL/min,柱温25 ℃,色谱柱的分离特性用保留因子k、分离因子α1,2、分离度Rs评价。
2 结果与讨论
2.1 手性固定相的元素分析
将色谱硅胶、8种手性固定相进行元素分析表征,结果见表1。与色谱硅胶相比,所制备的手性固定相的氮、碳、氢三种元素的含量都有所增加,说明替考拉宁和万古霉素已经成功键合到硅胶上。
2.2 手性固定相的扫描电镜(SEM)表征
将八种手性固定相做扫描电镜表征,结果如图1所示。从表征结果可以看出,八种手性固定相都是均匀的球形,符合液相色谱柱对固定相的要求,适用于高效液相色谱手性分离。

表1 元素分析测试结果

图1 八种手性固定相的SEM表征Fig.1 SEM Characterization of eight chiral stationary phase
2.3 手性固定相的红外表征
将空硅胶及八种手性固定相进行红外光谱分析,结果如图2所示。通过红外光谱图可知,四种替考拉宁固定相,在1 654 cm-1处酰胺羰基的特征吸收峰加强,结合元素分析说明替考拉宁已经键合到硅胶表面。四种万古霉素固定相,在2 900 cm-1左右均有C-H伸缩振动,结合元素分析结果,万古霉素和硅烷化试剂已经键合至硅胶表面上。
2.4 八种手性固定相拆分手性化合物
2.4.1 四种替考拉宁固定相手性拆分手性化合物
为了检测4种替考拉宁固定相手性拆分性能,对手性化合物进行拆分实验。以甲醇-水(85∶15,体积比,下同)作为流动相,测量波长为210 nm,此条件下用于拆分α-氨基酸;以甲醇-水(60∶40)作为流动相,测量波长为254 nm,此条件下用于拆分其他手性化合物,拆分结果如表2所示。
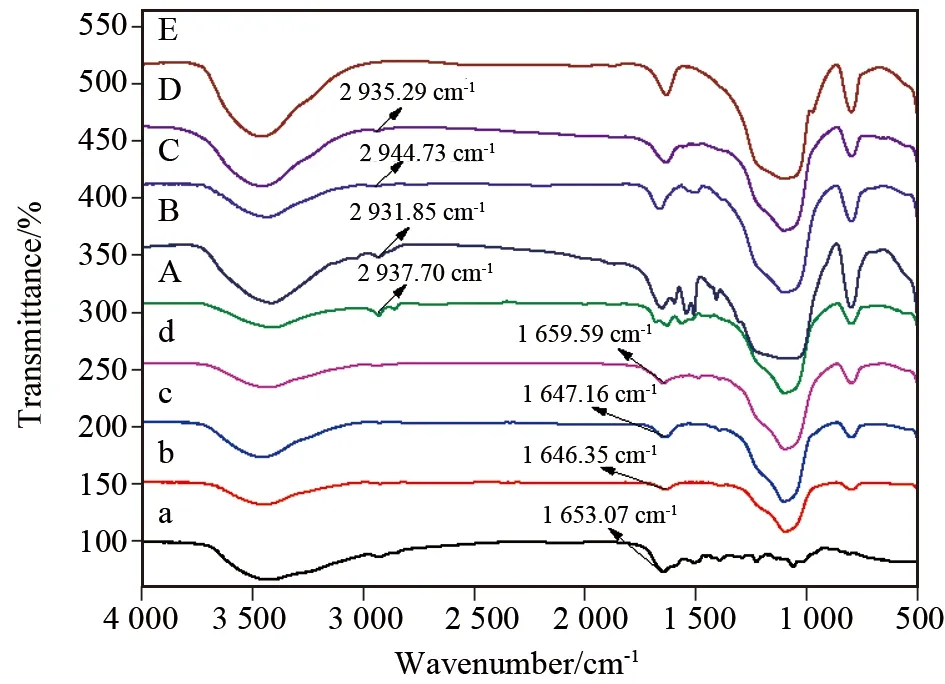
a.CSP-1 b.CSP-2 c.CSP-3 d.CSP-4 A:CSP-A; B:CSP-B; C:CSP-C; D:CSP-D; E:empty silica gel图2 空硅胶及八种手性固定相的红外表征图Fig.2 Infrared characterization of emptysilica gel and eight chiral fixed phase
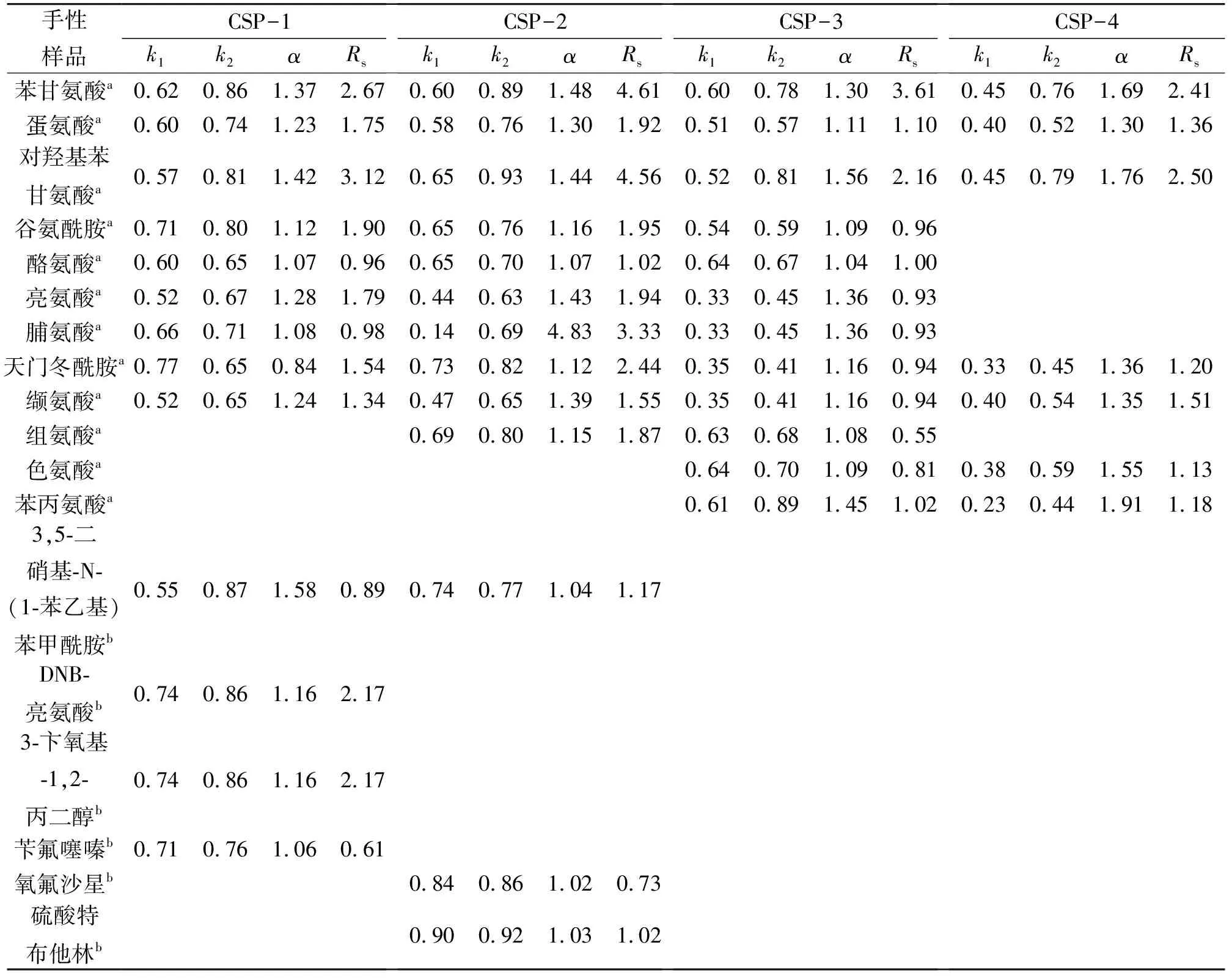
表2 CSP-1至CSP-4手性柱拆分手性化合物的结果
a代表流动相条件为甲醇-水(85∶15),b代表流动相条件为甲醇-水(60∶40)
从表2中的结果可知CSP-1成功拆分了13种手性化合物,对苯甘氨酸、蛋氨酸、对羟基苯甘氨酸、谷氨酰胺、亮氨酸、天门冬酰胺、DNB-亮氨酸这7种达到基线分离;CSP-2成功拆分了13个手性化合物,其中苯甘氨酸、蛋氨酸、对羟基苯甘氨酸、谷氨酰胺、亮氨酸、脯氨酸、天门冬酰胺、缬氨酸、组氨酸这9种达到基线分离;CSP-3成功拆分了9个α-氨基酸,其中苯甘氨酸、对羟基苯甘氨酸这2种达到基线分离;CSP-4成功拆分了7个α-氨基酸,其中对羟基苯甘氨酸、蛋氨酸、苯甘氨酸这3种达到基线分离。
图3为CSP-1至CSP-4拆分手性化合物的部分分离图谱。
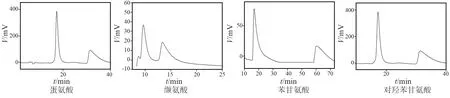
图3 CSP-1至CSP-4拆分手性化合物的部分分离图谱Fig.3 Partial separation map of CSP-1 to CSP-4 split chiral compounds
2.4.2 四种万古霉素固定相手性拆分手性化合物
为检测四种万古霉素手性柱对手性化合物的拆分效果,实验分别采用了甲醇-水(80∶20,体积比,下同)、甲醇-三乙胺(100∶0.03)、甲醇-三氟乙酸(100∶0.03)、甲醇-冰醋酸(100∶0.03)作流动相,流速0.1 mL/min,手性药物紫外检测波长为254 nm,氨基酸为210 nm测试,其分离结果见表3。
分离结果显示手性柱CSP-A拆分了8种手性化合物,其中氧氟沙星、扁桃酸、沙丁胺醇这3种手性化合物达到了基线分离,其余的5种手性化合物有不同程度的拆分;手性柱CSP-B拆分了9种手性化合物,其中噻吗洛尔、盐酸西替利嗪这2种手性化合物达到基线分离,其余7种手性化合物有不同程度的拆分;手性柱CSP-C拆分了5种手性化合物,其中1,2-二苯基-1,2-乙二醇、氨氯地平、二氢黄酮、酪氨酸这4种手性化合物达到基线分离,其余的手性化合物也有不同程度的拆分;手性柱CSP-D对5种手性化合物有不同程度的拆分。这四根手性柱在不同的模式之间有较好的互补性。万古霉素手性柱对流动相的组成以及酸碱的添加比例都比较敏感,如果能选择适当的模式,进一步优化这些参数,将会有更多的手性化合物被拆分开。
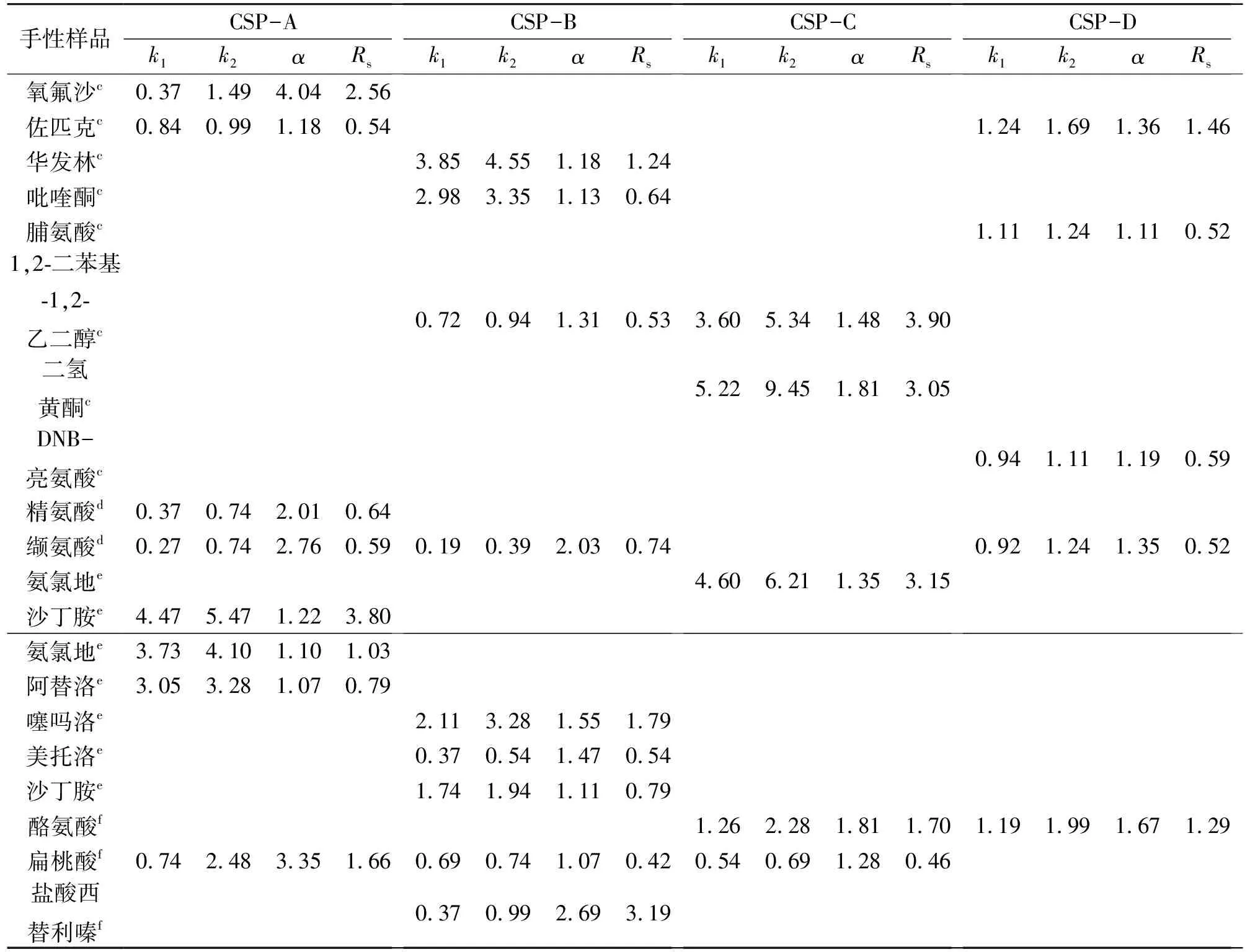
表3 CSP-A至CSP-D手性柱拆分手性化合物的结果
c代表流动相条件为甲醇-水(80∶20)、d代表流动相条件为甲醇-三氟乙酸(100∶0.03)、e代表流动相条件为甲醇-三乙胺(100∶0.03)、f代表流动相条件为甲醇-冰醋酸(100∶0.03),k1、k2代表保留因子、α代表分离因子、Rs代表分离度
图4是CSP-A至CSP-D拆分手性化合物的部分分离图谱,图谱如下所示:

图4 CSP-A至CSP-D拆分手性化合物的部分分离图谱Fig.4 Partial separation map of CSP-A to CSP-D split chiral compound
3 结论
所制备的八种手性固定相都表现出不同程度的手性拆分能力,特别是对α-氨基酸的拆分有较好的效果。四种不同的替考拉宁手性固定相相比较,CSP-1 在四种固定相中拆分的手性化合种类最多;CSP-2拆分的手性化合物个数较前者少,但是其对同种手性化合物的分离性能优于前者;其他2种固定相就拆分化合物的种类及拆分性能而言,都稍逊色于前两种固定相。替考拉宁手性固定相一共累计拆分了18种不同的手性化合物,其中有10种手性化合物达到了基线分离。四种不同的万古霉素手性固定相相比较,CSP-B 在四种固定相中拆分的手性化合种类最多;CSP-A拆分的手性化合物个数次之;CSP-C虽然只拆分了5种手性化合物,但拆分效果较好;CSP-D拆分效果相对不佳,但也有一定拆分效果。万古霉素手性固定相一共累计拆分了18种不同的手性化合物,其中有9种手性化合物达到了基线分离。所制备的八种不同手性固定相,键合臂不同,能拆分的对映异构体可能不一样,这些柱之间具有一定的互补性,在高效液相色谱法拆分手性化合物中有一定的应用前景。
