细胞焦亡与自噬的相互关系及其在动脉粥样硬化发生发展中的作用
2022-04-02潘天琦姚树桐
潘天琦 田 华,2 姚树桐
1.山东第一医科大学(山东省医学科学院)基础医学院,山东济南 250117;2.山东中医药大学中医学院,山东济南 250355
动脉粥样硬化(atherosclerosis,AS)是一种与脂质代谢紊乱相关的慢性炎症性疾病,严重影响人类的生活质量[1]。在正常组织中经常发生的细胞死亡,是维持机体正常生命活动所必需的,目前已经发现有坏死、凋亡、坏死性凋亡、焦亡、自噬和铁死亡等多种细胞死亡形式。细胞焦亡(pyroptosis)是机体炎症反应的重要组成部分,并参与AS 发生发展过程[2]。进一步研究细胞焦亡的特征、分子机制及其在AS 发病及防治机制中的作用,将有助于加深对细胞死亡方式和机制的认识,为AS 相关疾病的防治提供新思路,现就细胞焦亡在AS 发展中的研究进展作一简要综述。
1 细胞焦亡
1.1 细胞焦亡的特征
Cookson等[3]提出pyroptosis一词,即细胞焦亡,介于凋亡和坏死之间。细胞焦亡由caspase-1介导,其下游的炎症小体发生组装,由打孔蛋白Gasdermin家族的Gasdermin D(GSDMD)来执行。细胞焦亡时的形态学表现为细胞肿胀、染色质浓缩和DNA阶梯缺失,并且膜联蛋白Ⅴ(annexinⅤ)和TUNEL染色阳性。迄今为止,细胞凋亡被认为是可以被生命过程调控的死亡,坏死则被经典地描述为非调控细胞死亡[4-5]。
1.2 细胞焦亡的执行者—Gasdermin D蛋白
Gasdermin家族是一种在特定条件下表达的细胞质蛋白,也是焦亡的介质蛋白,主要包括GSDMA、GSDMB、GSDMC和GSDMD,其中GSDMD蛋白在细胞焦亡中的作用被广泛研究,其包含大约480个氨基酸,分为N端的Gasdermin-N和C端的Gasdermin-C两个结构域,通过一个灵活的连接子相连,Gasdermin-C结构域对Gasdermin-N 结构域起到抑制作用,全长GSDMD是无活性的,但这种灵活的连接子特别容易受到蛋白酶的攻击,可以作为一个简单的警报系统。当caspase-1/4接收到体内外传来的危险信号时发生活化,这些半胱天冬酶便可在GSDMD 的中心连接区进行切割,Gasdermin-N与Gasdermin-C结构域发生解离后可与真核细胞膜上的磷脂酰肌醇或原核细胞膜上的心磷脂结合,或与天然极性脂质混合物的脂质体特异性结合。Gasdermin-N通过电荷-电荷之间的相互作用发生寡聚化,形成直径12~14 nm的孔道,脂质体渗漏、生物膜溶解,细胞内外的渗透压被破坏,导致细胞肿胀并最终溶解,进而发生“细胞焦亡”[6-7]。
1.3 细胞焦亡的信号通路
1.3.1 经典焦亡途径 典型的炎症小体是由辅助蛋白和自我寡聚化支架蛋白组成的,它们通常属于核苷酸结合寡聚化结构域样受体(nucleotide-binding oligomerization domain-like receptor,NLR)家族,目前为止已经确定NLRP1、NLRP3、NLRC4和黑素瘤缺乏因子(absent in melanoma 2,AIM2)四个炎症小体。细胞焦亡作为一种炎症性死亡途径,在细菌、病毒、真菌等病原体相关分子模式(inflammatory pathogenassociated molecular patterns,PAMPs)和ATP、胆固醇等危险信号分子模式(damage-incorporated molecular patterns,DAMPs)的刺激下,在细胞质感应到PAMPs或DAMPs后,炎性小体进行组装并通过近距离诱导的自身蛋白水解激活炎性caspases-1,活化的caspase-1将关键的促炎IL-1家族细胞因子加工成引起焦亡的活性形式,但需要GSDMD介导其释放,IL-1家族还可以通过触发级联反应来放大炎症,激活NFκB、TNF-α等炎症细胞因子的分泌[8-10]。
1.3.2 非经典细胞焦亡途径 在发生感染的细胞胞浆中可以检测到细菌脂多糖(lipopolysaccharide,LPS)分子,LPS 激活小鼠体内caspase-11[11],研究发现caspsase-11/4/5 以高特异性和亲和力直接结合LPS,caspase-11 上的CARD 结构域可能是识别LPS的位点,多个CARD 肽段与LPS 结合形成了活化形式的caspase-1,经寡聚后被活化诱导焦亡,而CARD结构域缺陷时对LPS没有反应[12]。人体内并不拥有caspase-11,但是人caspase-4/5 与caspase-11 具有很高的同源性,因此caspase-4和caspase-5也具有相同的功能,这种功能代表了新的免疫识别模式。Caspase-11/4/5-LPS复合物被归类为非典型性炎症小体[13]。活化的caspase-11/4/5可以直接裂解GSDMD,在细胞膜上形成释放炎症介质的管道,大量炎症因子被释放,促进细胞焦亡[6]。见图1。
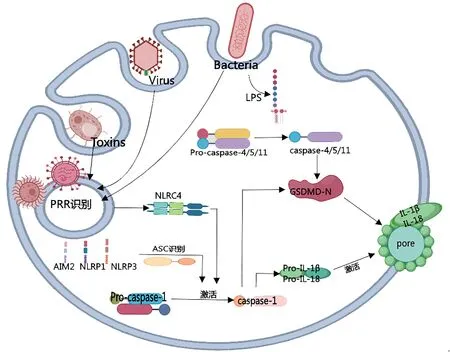
图1 细胞焦亡的信号通路
Caspase-1 基因敲除小鼠对注射致死剂量的LPS 具有高度抵抗力,同时发现缺少caspase-11 的小鼠也具有此特性[14],这一研究发现已显示出细胞焦亡的重要作用,但为了开发防治AS的新策略,需要对二者的潜在关系进行更深入的了解。
2 细胞焦亡与AS的关系
AS 是一种慢性炎症性疾病,血管内皮细胞(vascular endothelial cell,VEC)、平滑肌细胞(vascular smooth muscle cell,VSMC)及巨噬细胞等多种细胞参与AS 的发生发展,同时焦亡也可以发生在这些细胞中,而焦亡作为细胞死亡的重要形式之一,是针对细胞感染产生的一种炎症反应。对颈动脉内膜切除术患者的AS 斑块进行检测,发现焦亡相关蛋白的表达均上调,且这些蛋白在稳定斑块中的表达明显低于不稳定斑块[15],说明细胞焦亡与AS斑块稳定性密切相关。
众所周知,VECs 功能障碍和死亡是AS 发生发展的始动环节。活性氧(reactive oxygen species,ROS)和氧化低密度脂蛋白(oxidized low density lipoprotein,ox-LDL)等都是NLRP3 的直接激活剂,而且都可以导致VECs 发生损伤[16]。新近研究表明,琥珀酸作为三羧酸循坏的中间代谢产物,可以增加VECs 中ROS 含量,导致焦亡相关蛋白的表达明显上升[17],在AS 进展中起着重要作用。文献报道甲基莲心碱作为自由基清除剂,可增强超氧化物歧化酶活性,抑制ROS 的产生,从而抑制NLRP3 的激活和炎症因子释放,减轻内皮细胞焦亡,并可减轻AS 病变[18]。另一篇报道中,ox-LDL 可以通过上调miR-125a-5p 和下调甲基胞嘧啶双加氧酶(tet methylcytosine dioxygenase 2,TET2)诱 导VECs 焦亡,因此,靶向miR-125a-5p/TET2 通路可能调节DNA 甲基化,改善线粒体功能,抑制焦亡,是AS 潜在的表观遗传生物学标志物和治疗方向[19]。同时,胰高血糖素样肽1(glucagon-like peptide-1,GLP-1)也是通过减轻ox-LDL诱导的VECs焦亡来发挥其抗炎抗氧化作用的[20]。镉(Cd)和丙烯醛是常见的环境污染物,与AS相关性心血管病相关,研究报道其可能是通过NLRP3途径激活caspase-1,诱导内皮细胞焦亡,而抑制NLRP3则可减轻内皮细胞焦亡[21]。以上大量研究证实,对VECs焦亡通路进行干预,可减轻焦亡,可能成为AS防治的重要措施。
VSMCs 是AS 病变中主要的细胞成分,被认为在心血管疾病的发展中起到至关重要的作用[22],近年来研究表明VSMCs焦亡与AS斑块易损性密切相关。Ox-LDL 通过激活caspase-1 炎症小体途径导致VSMCs焦亡,造成胶原和基质丢失、纤维帽变薄,极易导致斑块破裂,坏死性粥样物质进入循环血液,从而导致动脉血栓形成和急性血管事件的发生[23]。经过caspase-1抑制剂VX-765干预后发现载脂蛋白E基因敲除小鼠的VSMCs 焦亡程度显著减轻,并可通过降低主动脉中脂质负荷而显著抑制小鼠AS的发展[24]。血小板源性生长因子(platelet-derived growth factor,PDGF)是一种常见的肽类调节因子,有利于VSMCs 等细胞生长。也有研究发现,经PDGF 处理后VSMC 中NLRP3、caspase-1 表达均上调,提示PDGF能诱导VSMCs中NLRP3炎症小体活化,引起VSMCs焦亡[25]。
巨噬细胞焦亡在AS 病变中起到关键性作用。Shi等[26]观察到在颈动脉内膜切除术患者的AS斑块中NLRP3、ASC、caspase-1、IL-1β 和IL-18 表达均上调,且其表达主要见于不稳定斑块的晚期AS病变,以及巨噬细胞和泡沫细胞的细胞质中,增加了斑块脆弱性。本课题组既往研究结果显示,晚期糖基化白蛋白(advanced glycated albumin,AGE-alb)是AS的一个重要致病因素,可浓度依赖性地增加巨噬细胞焦亡,而给予MCC950(NLRP3 特异性抑制剂)处理后焦亡程度明显减轻[27]。胆固醇作为AS形成的主要原因,能够触发NLRP3、caspase-1 信号途径的激活,诱导巨噬细胞焦亡,可能是AS中炎症形成的关键因素。以上研究说明,细胞焦亡与AS 的诱因息息相关,这些诱因可以激活炎症小体,导致慢性炎症,进而导致AS。炎症小体是把双刃剑,在急性疾病中炎症小体被激活后有助于清除死亡细胞并启动组织修复,而在慢性疾病中炎症小体的持续激活是有害的。因此为了探索炎症小体作为治疗靶点的潜力,需要对介导炎症小体激活的有益和有害作用的信号分子进行更多的探索。
3 细胞焦亡与自噬的关系及其在AS进展中的作用
自噬(autophagy)是一个古老的、进化上保守的分解代谢过程,与细胞质产物的降解和循环密切相关,它可以将细胞质货物运输到溶酶体,并通过溶酶体水解酶进行后续降解。自噬缺陷可以导致包括AS在内的多种疾病,因此了解自噬调节机制及其与AS的关系对于开发新的AS治疗方法至关重要。近年来研究表明,自噬与细胞焦亡存在相互作用,并参与AS发生和发展。Van等[28]认为自噬功能障碍可以激发单核细胞进行线粒体介导的NLRP3炎症体激活,从而导致IL-1β的过度分泌。在焦亡通路中,PRR是感知PAMPs 和DAMPs 外源性和内源性“危险”信号的传感器,可以通过微管相关蛋白1 轻链3(microtubuleassociated protein 1 light chain 3,LC3)或beclin-1 信号通路调控自噬,而自噬能够对PRR 和NLRP3介导的炎症形成负反馈调节,即抑制其组装活化,以维持体内平衡,防止发生过度炎症反应[29]。小檗碱(berberine,BBR)可以通过上调巨噬细胞的自噬水平抑制NLRP3的激活,而降低beclin-1水平或添加自噬抑制剂后则可拮抗BBR对NLRP3生成/表达的抑制作用[30]。来自对褪黑素(melatonin,MT)的研究表明,MT具有抗炎特性,用其治疗高脂饮食诱导的ApoE-/-小鼠,发现AS中NLRP3活化和IL-1β分泌均减少,而这种保护效果可被自噬抑制剂3-甲基腺嘌呤所逆转,这表明MT通过其抗炎特性抑制AS的进展,另外观察到MT可以通过消除巨噬细胞中ROS的同时降低NLRP3炎症小体的表达,最终MT治疗后发现AS斑块明显减小且稳定性增加[31]。用活性氧抑制剂治疗巨噬细胞时发现NLRP3 炎症体的表达显著降低,为了验证自噬在这一过程中的作用,巨噬细胞用自噬抑制剂3-甲基腺嘌呤(3-methyladenine,3-MA)或诱导剂雷帕霉素处理,用3-MA处理后NLRP3表达明显上升,而雷帕霉素处理后NLRP3炎症体的表达被显著抑制。这种方式证实了ROS、自噬和NLRP3炎症小体激活之间的关系[32]。当NLRP3被过度激活时,亦会抑制自噬。在LPS 诱导的抑郁大鼠中,NLRP3被过度激活后引发神经炎症,而beclin-1水平和LC3-Ⅱ与LC3-Ⅰ的比值显著降低,提示自噬受到显著抑制[33]。也有文献报道自噬可正向调节酵母菌中NLRP3炎症小体的激活[34]。当细胞处于饥饿状态时,自噬可以通过Atg5依赖性途径增强caspase-1和炎症小体活化,炎症因子释放增加,加重组织的炎症性损伤,根据目前研究来看这种现象可能是酵母菌独有的,而哺乳动物细胞中细胞焦亡与自噬之间的确切关系及其在AS不同阶段的具体作用还有待进一步研究。
临床研究发现,IL-18 在人体AS 斑块中高表达[35],因此IL-1β在AS发展中的关键作用突出了这种细胞因子作为干预的潜在目标,Canakinumab抗炎治疗后血栓形成结果研究试验提供了强有力的支持,患者采用IL-1β阻断治疗后,显示出预防心肌梗死复发的效果[36]。动物研究表明,IL-1β 阻断治疗后,ApoE-/-小鼠中已经形成的AS斑块显著减少[37]。但IL-1β被阻断后可能会干扰感染免疫反应,导致严重的感染,Li等[38]已经解决此问题,试验中采用人造血小板微颗粒(platelet microparticles,PMs)为载体,将抗IL-1β抗体输送到心脏,并在心脏损伤的治疗中达到预期的结果。
4 自噬可能成为治疗AS的重要靶点
研究发现,炎症过程和脂质代谢共同促进了动脉壁AS斑块的形成,通过阻断相关分子(例如NLRP3、caspase-1、IL-1β/18)的诱导因素(如ox-LDL)可以抑制焦亡对AS进展的影响[39]。焦亡是一种广泛发生的炎症反应,随着研究的深入这种死亡模式逐渐被阐明,但仍有一些问题有待解决,如除自噬外还有何因素调节焦亡,Gasdermin家族其他成员在焦亡中发挥什么作用,在AS的发生发展中扮演什么样的角色。
鉴于细胞焦亡与AS 之间关系密切,那么干扰特定的细胞焦亡信号通路,一定程度上可抑制AS的发生发展。近年来,大量研究集中在自噬和炎症小体之间的关系上,适度的激活自噬可以抑制炎症小体的活化,且自噬抑制炎症小体的理论已经在许多炎症性疾病的治疗中得到试验证明,但是二者之间的关系尚未完全阐明[40]。因此我们可以对这一方向进行深入探索,不仅有助于研究者和临床医生了解自噬和炎症的相关病理生理过程,还可以为AS靶向药物的开发提供试验依据。
利益冲突所有作者均声明不存在利益冲突
