飞行时间二次离子质谱在锂基二次电池中的应用
2022-03-23赵志伟彭章泉
赵志伟,杨 智,彭章泉
(中国科学院大连化学物理研究所,辽宁 大连 116023)
能源和环境是驱动人类社会可持续发展的重要源动力。为了社会的可持续发展,必须改变现有的能源生产和消费方式以避免灾难性后果,如化石燃料的过度使用导致的气候变化、资源枯竭等。电化学储能装置由于其经济、绿色、转化效率高等优势吸引了学术界和产业界的广泛关注,其中最成功的案例便是锂离子电池[1]。自1990年锂离子电池被日本索尼公司商业化以来,已经彻底改变了人们的日常生活并主导了便携式能源消费市场。但是,随着不断增加的社会能源需求,如长续航电动汽车和规模化能源储存(如太阳能和风能等),锂离子电池技术受限于有限的能量密度提升空间,已经开始呈现出发展疲态。因此,锂离子电池迫切需要技术革新来达成高功率密度、高安全性和长寿命的特点以满足未来不断升级的能源需求[2-3]。同时,最近几年兴起的高比能电化学储能技术,如锂硫电池[4]和锂氧电池[5-6],也开始逐渐受到人们的广泛关注。然而,这些新兴的电化学储能技术在实际应用中仍然面临着诸多挑战,比如较差的循环寿命、倍率性能以及严重的副反应等[7-10]。
为了革新和最大化锂基电池(包括锂离子电池、锂硫电池和锂氧电池)技术潜力以满足未来不断升级的社会能源需求,它们的电极材料、电解液等关键电池组分需要优化、创新和突破。为此,需要深入理解电极/电解液界面电化学反应机制,这是因为界面电化学反应决定了电极材料和电解液的衰退、离子传输及电荷传递等过程并最终影响器件的电化学性能。然而,直接分析锂基电池电极/电解液界面过程充满了挑战性[11-13]。这是因为电池界面反应空间尺度小、时间尺度短、中间体/产物(气/液/固)复杂多变。在过去的几十年里,大量的非原位技术,如X射线技术、红外光谱、核磁等,被广泛应用去探究电池界面过程的原始态及循环后状态,并获得了丰富的形貌、结构和化学信息,极大地推动了锂基电池的深入发展[14]。通常来说,非原位手段经常忽略短寿命中间体,很难获知电化学界面的动态演变过程,因此电化学反应机制经常难以得到充分理解。随着科学仪器以及智能制造技术的不断发展,将传统电化学和先进表征分析技术结合(如原位电镜、核磁、X-射线技术),电化学反应过程被探测的时空分辨率不断提高,能量分辨率不断拓宽,为理性设计高效电极材料并优化电池性能提供了充分的技术支持[15-19]。其中,飞行时间二次离子质谱(time-of-flight secondary ion mass spectrometry,ToF-SIMS)是一种质量分辨并兼具时空分辨的技术。传统的电化学研究手段与ToF-SIMS结合,能够快速直接原位(ms级)鉴定电化学反应微量的中间体/产物(ppm水平甚至更低浓度,1ppm=10-6),并同时关联化学信息和电极/电解液界面空间信息,为揭示完整的电化学界面反应物理化学图像提供了可能性[20]。
本综述首先简要介绍了ToF-SIMS技术原理以及和传统电化学之间的联用技术。然后,总结了ToF-SIMS技术在锂基电池(包括锂离子电池、锂硫电池和锂氧电池)中的实际应用,并重点讨论了该技术如何去帮助研究人员了解电池过程并设计更好的电化学储能系统。最后,阐述了ToF-SIMS技术在锂基电池中的挑战和机遇。
1简要概况
SIMS技术是指使用脉冲初级离子束(Bin+、Au+、C60+、Arn+等)撞击样品表面,其结果是从样品表面的前几个单层中产生次级离子(+/-)、电离原子和分子,然后溅射进入质谱仪中进行分析(图1)[21-22]。SIMS分析可用于分析样品表面某位置的成分,也可以构建样品表面二维或三维成分分布图。SIMS电离技术分为“静态”和“动态”SIMS。其中,“静态”SIMS是指使用少量初级离子(通常<1013ion/cm2)的分析,该剂量仅去除样品表面1%的原子位点。相比之下,“动态”SIMS是指去除相对大量原子的SIMS技术。由于样品分子碎片之间的化学键在少量初级离子条件下更有可能保留,“静态”SIMS技术因此可以用于获取分子信息,而“动态”SIMS技术不保留化学键,可用于提供样品中元素分布的三维数据[23]。
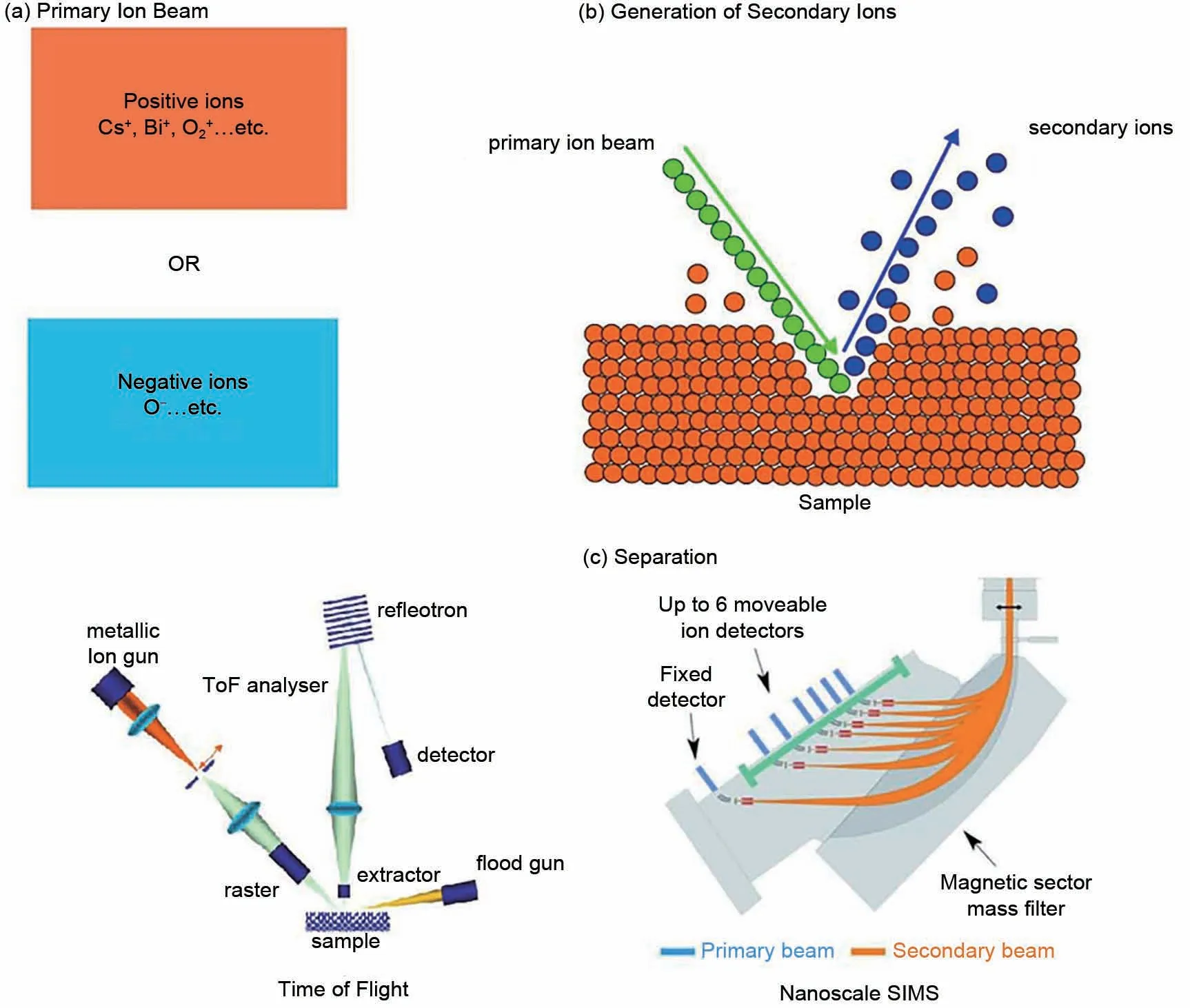
图1 二次离子质谱(SIMS)分析原理:(a)SIMS一次离子源;(b)一次离子束在溅射过程中撞击样品表面,产生二次离子;(c)二次离子通过飞行管或四极杆实现分离[23]Fig.1 Principles of secondary ion mass spectrometry(SIMS)analysis:(a)SIMS primary ion beam:positive ions or negative ions;(b)the primary ion beam strikes the sample surface during sputtering and then produces secondary ions;(c)secondary ions are accelerated towards the detector,either through a flight tube(ToF-SIMS)or through magnetic separation using a quadrupole(NanoSIMS)[23]
虽然所有SIMS技术原理相同,但它们在初级离子源、离子束撞击样品的电压、移除的样品量以及用于检测的离子分离方面略有不同。最常见的SIMS仪器类型是ToF-SIMS和NanoSIMS。ToFSIMS是一种灵活的技术,“静态”ToF-SIMS为了获取空间分辨率需要更高浓度的目标分析物样品,也可以控制初级离子束的电流或类型,选择性从分子碎片或原子信息中获取信息。ToF-SIMS技术使用飞行时间作为检测模式,用初级离子源溅射产生带电的次级离子,然后加速进入飞行室,随后所测试离子加速通过飞行室并根据离子到达时间进行分离到达检测器。在典型的双束ToF-SIMS仪器中,初级离子枪用于产生次级离子进行分析,而第二个“溅射”离子枪用于深度剖析[24]。
相比之下,NanoSIMS不能在“静态”模式下运行。相反,NanoSIMS始终在“动态”模式下运行[25]。NanoSIMS只能生成原子尺度的次级离子。因此,在没有分子二次离子的情况下,NanoSIMS中的同位素测量可用于推断分子分布。NanoSIMS方法通常使用Cs+源(空间分辨率可达50nm)增强负二次离子的电离以及O-/O2-源增强正二次离子的电离。NanoSIMS的测试离子被偏转到六个可移动的检测器和一个固定的检测器,这样在每次分析期间总共可以检测到七种物质。因此,NanoSIMS仪器的限制是一次最多测定七个物种,且无法同时分析正负二次离子,仅能提供小分子碎片分布的信息。因此,ToF-SIMS技术使用在能源电化学领域更为广泛一些,后文主要介绍ToF-SIMS在锂基电池中的应用。
能源电化学领域最常见的界面是固液界面。电极表面原子具有与电极本体完全不同的化学环境。因此,电极表面的原子和电子结构往往表现出高的化学/电化学反应活性。因此,理解电极/电解液界面过程(如电荷传输、界面膜生长等)对于增强储能器件性能至关重要。ToF-SIMS技术的出现,为研究电极表面过程提供了更多的可能性。不同于其他表征技术,ToF-SIMS可以将电极表面的化学信息和空间信息直接关联。然而,ToF-SIMS测试需要高度真空环境,难以直接应用于电化学的固液反应界面。尽管非原位实验可以提供电化学过程中电极始末态的信息,但将电极从电解液中转移到真空中可能会存在一些问题,而且也难以获取电极反应中间态信息。因此,实现ToF-SIMS技术与电化学的联用具有重要意义。
在2014年,Liu等[26]利用微流控技术集成了三电极系统,首次使用ToF-SIMS技术原位测试了电极/电解质界面过程。其主要原理是将工作电极材料溅射到氮化硅(SiN)薄膜上,SIMS一次离子源轰击SiN,穿透SiN薄膜后轰击电极/电解液界面,溅射出的界面二次离子被检测器检测。值得注意的是,微流控池体孔径较小(<2μm)时,表面张力可以将液体保持在微流控池体中而不会在高真空条件下(<5×10-7mbar,1bar=0.1MPa)喷出。图2(a)~(c)是电化学微流控池体示意图,图2(d)是微流控模型电池的数码照片,图2(f)是ToF-SIMS样品台上的微流控电化学池。在测试过程中,与真空兼容的电泵使得通道中电解液持续流动。微流控池体可以通过重新填充不同电解液来多次使用。随着实验的进行,ToF-SIMS初级离子束所钻孔会越来越大最终难以维持测试所需真空度,因此原位电化学ToFSIMS联用技术并不能长时间在同一位置采集数据点,实验时需要格外小心。
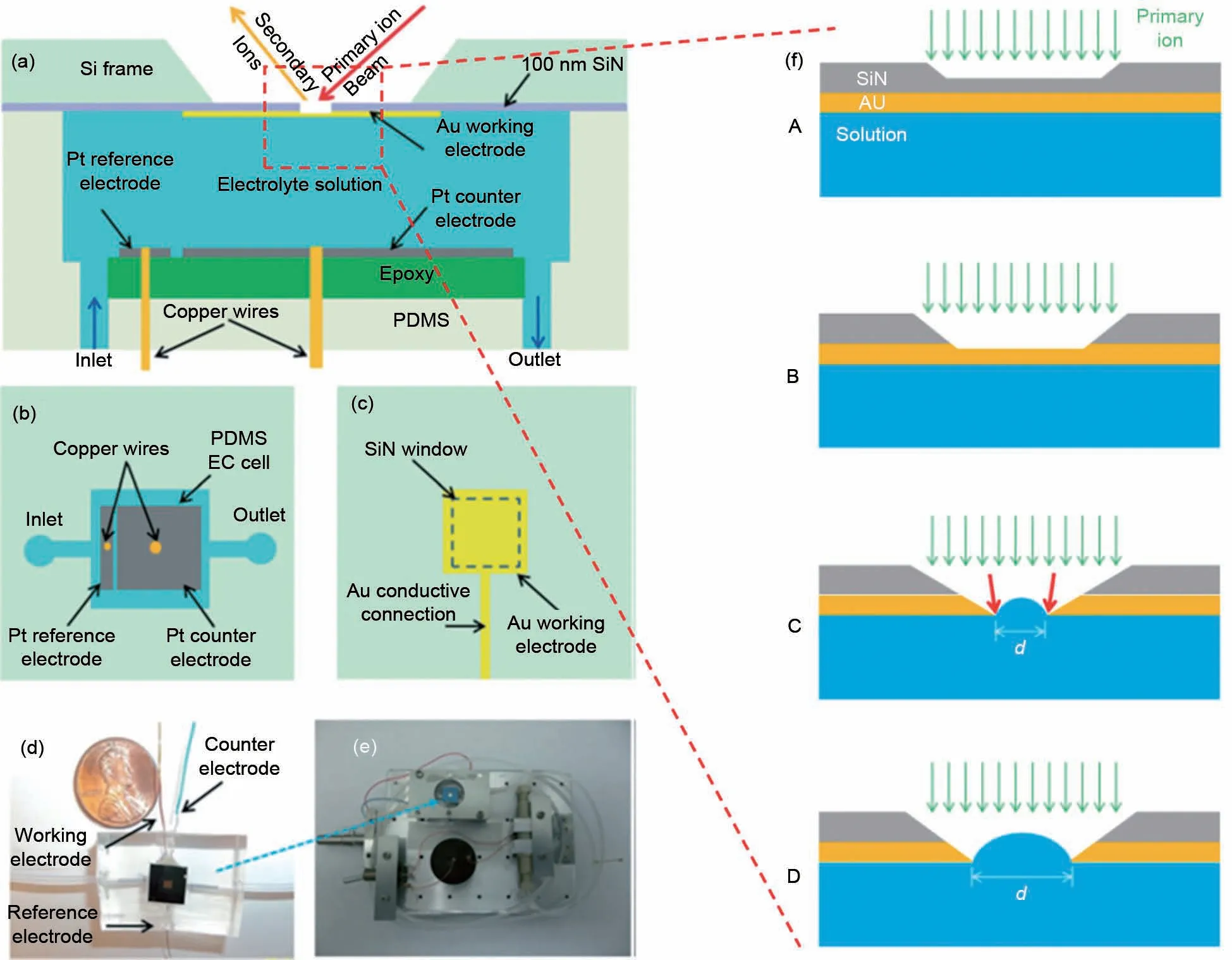
图2 首台原位电化学ToF-SIMS分析装置示意图,包括(a)侧视图示意图;(b)Pt对电极和参比电极的顶部透视图;(c)Au工作电极的顶部透视图;(d)装置数码照片;(e)组装在ToF-SIMS样品台上的电化学组件;(f)原位测量过程SiN孔径演变示意图[26]Fig.2 The first in situ electrochemical ToF-SIMS analysis device,including(a)side view,(b)top view of the Pt counter electrode and reference electrode,(c)the top view of the Au working electrode,(d)the photograph of the device and(e)EC-cell assembly on the ToF-SIMS stage;(f)a schematic illustration of the aperture evolution during in situ measurements[26]
2 应用介绍
2.1 锂离子电池
锂离子电池是当今移动电子设备的主流电源。在过去的几年,锂离子电池在电动汽车、规模储能方面的应用发展势头迅猛。然而,锂离子电池在三十多年发展历史中仍然有诸多的基础问题未得到充分解决[27]。
2.1.1 溶剂化结构
锂离子溶剂化结构是一个充满争议性和挑战性的主题。这是因为,锂离子电池溶剂化结构不仅影响本体电解液中Li+的传输,而且也会对界面固体电解质膜(SEI或CEI)的化学组成、结构、锂离子脱溶剂化动力学产生显著影响。研究者广泛认可的是,Li+与溶剂分子能够发生配位作用,但配位数是多少(是否完全溶剂化或形成离子簇),阴离子与溶剂和Li+如何相互作用,仍在争论中。当电解液由多种电解质盐和溶剂组成,并且浓度、温度等因素改变时,这些问题将变得更为复杂。
先进的实验方法(如红外光谱、拉曼光谱、核磁等)和理论计算(密度泛函理论、分子动力学模型)已经被广泛应用于研究Li+与电解液的相互作用[28-31]。然而,这些实验技术只能提供“化学位移”信息,即溶剂分子化学环境的变化,同时理论计算结果也难以通过实验方法验证,其可靠性亟需证明。因此导致溶剂化结构争议性的本质问题是无直接的分子证据。2012 年,Xu 等[32]使用电喷雾质谱(ESI/MS)成功确定了溶剂化物质和溶剂化壳的组成,指出Li+优先于碳酸丙烯酯(PC)和碳酸乙烯酯(EC)配位,成功解释了EC 对于锂离子电池石墨界面化学的重要性。尽管ESI/MS能够提供溶剂化结构的重要信息,但是其仍然存在一些固有挑战,例如ESI/MS 仅适用于极稀盐溶液,电喷雾过程不能直接暴露于空气(锂离子电池绝大多数电解液对水分敏感)等。
2018 年,Zhang 等[33]利用ToF-SIMS 方法,获得了电解液的直接溶剂化结构信息,为Li+溶剂化作用的基础研究提供了新的方法与视角。如图3(a)所示,使用氮化硅(SiN)薄膜将电池中的液体电解质与SIMS 真空隔开。在ToF-SIMS 分析过程中,使用聚焦的初级Bi3+离子束在SiN 膜上钻出一个小孔(直径约2 μm),随后电解液的质谱信号被收集。该设计沿用了原位电化学ToF-SIMS 联用技术的微流控设计,可以在任何商业SIMS 仪器中使用。由于无电喷雾过程,分析的电解液类型也无限制。最重要的是,通过控制离子束的脉冲时间和强度,该设计不仅可以用来检测本体溶剂化结构,也可用于电极/电解液界面双电层区域的分析(下一节将介绍)。Zhang 等[33]利用该技术成功分析了LiPF6/EC-DMC电解液(典型锂离子电池电解液配方)的溶剂化结构。如图3(b)展示的正离子模式质谱图,含EC 物种{[Li+EC]+(m/z=95),[Li+(EC)2]+(m/z=183)和[Li+(EC)3]+(m/z=271)}的信号强度比含DMC 的物种{[Li+DMC]+(m/z=97),[Li+EC+DMC]+(m/z=185)和[Li+(EC)2+DMC]+(m/z=273)}强2 个数量级以上,表明EC优先于Li+发生配位。这些直接的实验证据能够很好解释电解液在石墨表面还原形成的SEI产物来自于EC 而不是DMC。先前研究经常忽略锂离子电池电解液中阴离子-溶剂的相互作用,通常认为电解质盐的阴离子的溶剂化比Li+弱得多。在1.0 mol/L的LiPF6/EC-DMC电解液负离子质谱图中[图3(c)],能够清楚观察到溶剂化阴离子的峰[PF6+EC]-(m/z=233)也强于[PF6+DMC]-(m/z=235),表明阴离子也会与EC 发生优先溶剂化作用。该结论进一步支持EC 比DMC 更容易在石墨表面还原的结论,同时该研究也提醒研究者阴离子-溶剂相互作用可能对于SEI(如阴离子分解形成富含无机物的SEI)的组成结构起关键作用,需要进一步探索。
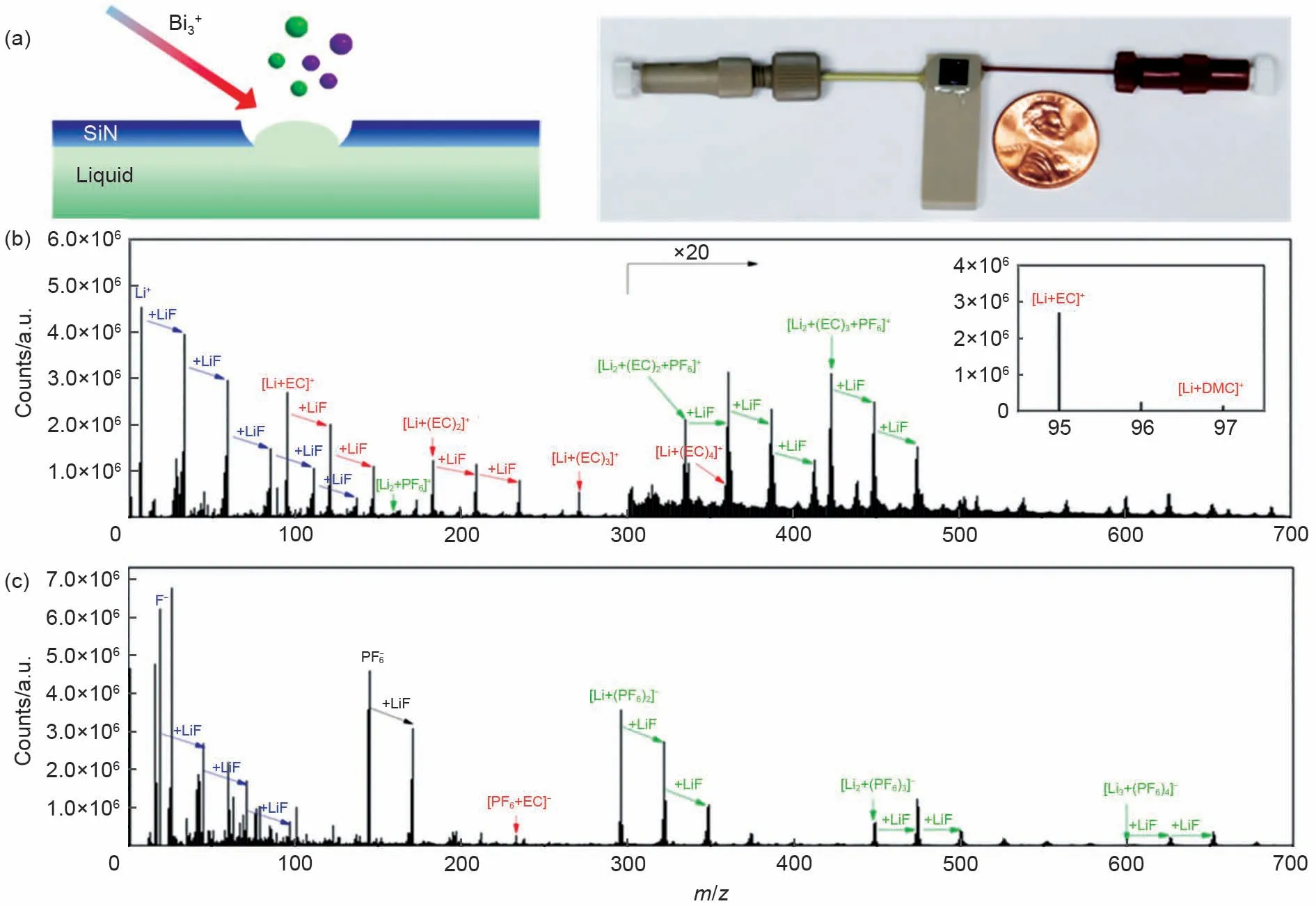
图3 (a)SIMS测试装置示意图及真空兼容的液体电池数码照片;(b)1.0mol/L LiPF6/EC-DMC电解液的正离子模式质谱图;(c)1.0mol/L LiPF6/EC-DMC电解液的负离子模式质谱图[33]Fig.3 (a)schematic illustration of SIMS measurement and the photograph of the liquid battery that is vacuum compatible;(b)positive ion spectra of1.0mol·L-1LiPF6in EC-DMC electrolyte;(c)negative ion spectra of 1.0mol·L-1LiPF6in EC-DMC electrolyte[33]
2.1.2 SEI形成过程
如2.1.1 节所述,电解液溶剂化结构在SEI 形成过程中扮演关键角色。通常认为,溶剂化的Li+接近带负电的电极表面,而阴离子被施加的负电势所排斥,导致阳离子和阴离子界面双电层的重排。双电层内组分优先还原形成SEI。因此,双电层结构和组成对于锂离子电池SEI化学和最终的电池性能将产生重大影响。然而,大多数研究经常从本体电解液溶剂化结构去预测SEI 化学,电极界面与SEI化学之间的联系缺乏直接的实验证据。
2020年,Zhou等[34]使用原位电化学ToF-SIMS联用技术提供了电极/电解液界面复杂过程的准确动态分子信息。该研究以Cu|1.0 mol/L LiTFSIDME|LiCoO2体系为模型[图4(a)],使用Si3N4膜将模型电池与高真空隔开。如图4(b)所示,在SIMS分析的初始阶段,仅检测到Si3N4相关信号,在Bi3+初级离子束穿过Si3N4膜后开始出现界面信号,在初级离子束穿过界面层后检测到本体电解液信号。在此过程中,由于电解液的高流动性,初级离子束无法钻入电解液。需要说明的是,所溅射的Cu纳米离子(30~40nm)是多孔结构,Bi3+初级离子束穿过Si3N4膜后,Cu+和界面二次离子信号同步出现。如图4(c)所示,在开路状态时,电极界面正负电荷均匀分布,阴离子(FSI-)和阳离子(Li+,Li-DME+)保留在Cu电极表面;在充电至1.0V,电场作用力下溶剂化的Li+吸附在Cu电极表面,FSI-浓度降低(见文献[34]);充电至2.0V时SEI在电极/电解液界面处形成,靠近Cu电极表面,6Li+出现在[Li-DME]+、OCH3-和FSI-信号之前,这表明形成了含锂SEI(内部SEI)。放电返回开路状态,结果与充电至2.0V时情况相似,这表明放电后内部SEI层没有溶解。然而,放电后OCH3-信号变得非常强,这表明除了内部SEI层之外还形成了有机富集层(外部SEI层)。上述观察结果表明,一旦电极带负电,负电荷电极不利于阴离子还原,SEI化学组成实际上由溶剂分子主导,导致内部SEI本质上是薄的、无机的和致密的内层,主要是溶剂还原形成的Li2O。形成内层后,出现富含有机物的外层,电解液可在其中扩散渗透。该技术实现了电极/电解液界面化学的原位观察,将有助于设计更好的锂离子电池界面化学。
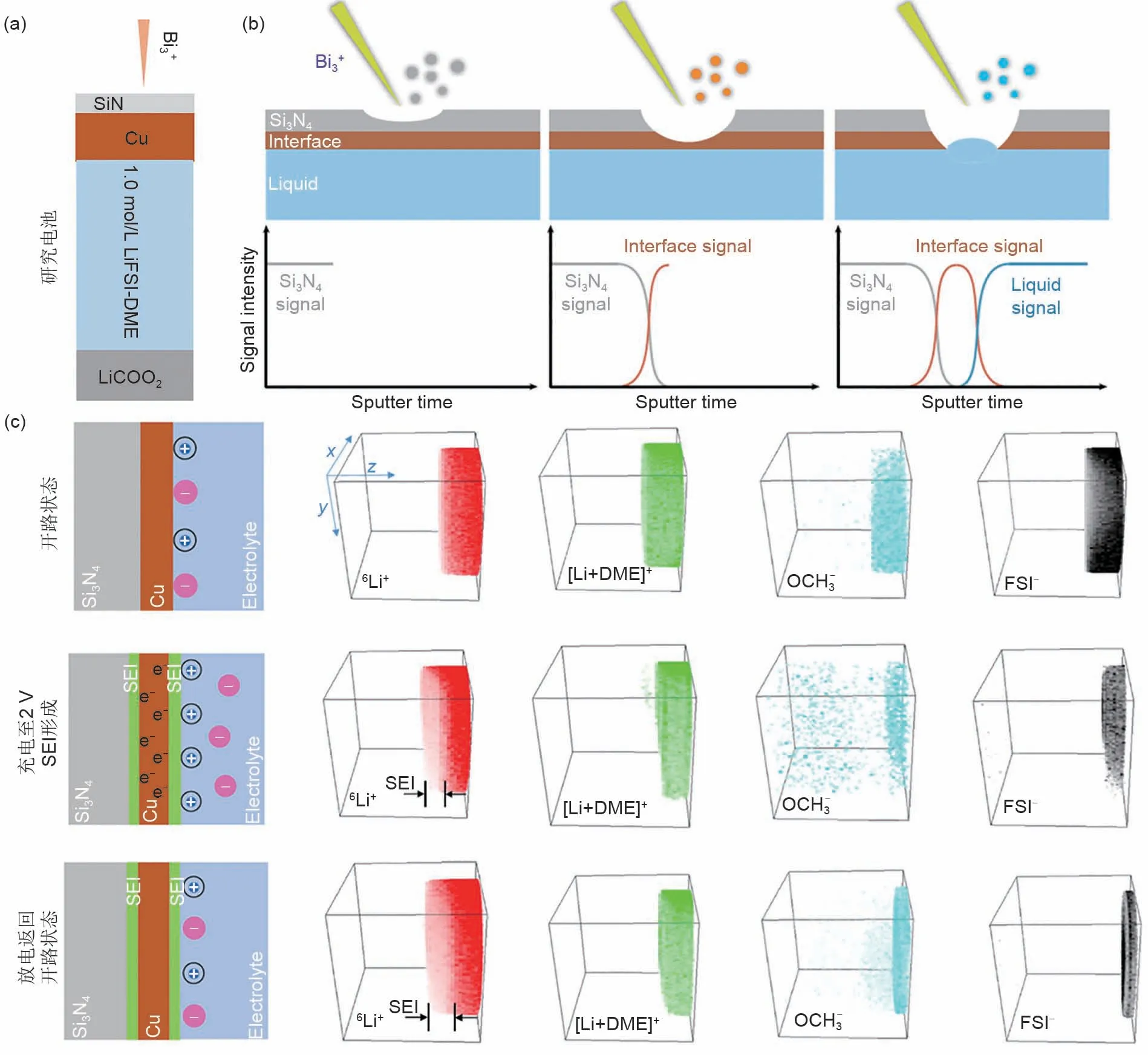
图4 (a)模型电池示意图;(b)原位ToF-SIMS界面分析示意图;(c)Cu电极表面重要二次离子物种在开路状态,充电至2V,放电返回开路状态的三维构建图像[34]Fig.4 (a)schematic of the model cell;(b)a schematic illustration of in situ ToF-SIMS analysis of solid-liquid interface;(c)3D reconstructions of the important SIMS signal on OCP state,charging to2.0V and discharged OCP state on Cu electrode surface[34]
2.1.3 SEI生长过程
2.1.2节介绍了锂离子电池中SEI是如何形成的,本节介绍SEI如何动态生长。在先前的研究中,光谱学方法能够给出SEI平均的化学信息,AFM或电镜技术能够提供SEI形貌演变但很少的分子信息,因此SEI的生长过程或结构演变仍然不清晰。作为对实验表征的补充,在不同尺度上进行了各种方法建模,如密度泛函理论计算、蒙特卡罗和分子动力学模拟等。但是,时间和空间的限制使得模拟仅关注非常短跨度的局部SEI形成过程。
Ushirogata等[35]提出SEI形成初始阶段的“溶液介导”机制,电解质分解产物将首先解吸并形成聚集体,随后吸附到电极表面以形成SEI初始层。Takenaka等[36]提出“自上而下”的SEI生长过程,即新的SEI层形成在现有层的顶部。然而,该模型假设整个SEI形成过程中反应速率恒定,简化的反应速率限制了其预测准确性。
Liu等[37]利用同位素辅助的ToF-SIMS技术,揭示了连续时间尺度上的SEI生长动力学,特别是电解液初始分解过程SEI的形成。图5(a)所示为所使用的模型电池,即Cu|6Li+-电解液|7Li。当电解液还原在Cu电极表面形成SEI后,6Li+优先参与SEI形成,7Li负极提供7Li+并补充到电解液中,因此,SEI在生长过程中,电解液中6Li∶7Li的比例会随着时间的推移而降低,即SEI生长的时间尺度可以表示为SEI中的6Li∶7Li比例。最初形成的SEI层的6Li∶7Li比例应高于随后形成的SEI部分[图5(b)]。利用同位素标记技术,在SEI生长过程完成后,也可以提取SEI生长动态信息。图5(c)和5(e)是电池放电后SEI表面溅射二次离子随SIMS溅射深度变化的函数。在其深度剖面中,深度0是指电解液/SEI界面处SEI的最上表面,更大的深度数表示SEI中靠近Cu表面的位置。结合Cu+和SEI的其他组成代表物种(即C2H2-和Li2F-)的强度变化趋势,SEI/Cu界面位置在30nm深度左右。图5(c)中,电解液/SEI界面处可以观察到较高的6Li∶7Li的比例,相反,较低的6Li∶7Li的比例在SEI/Cu的界面附近,表明最初形成的SEI靠近电解液侧,最后形成的SEI靠近电极侧。因此,SEI的生长在空间上遵循“自下而上”的顺序,即新产生的SEI在SEI/电极界面处形成,并随着SEI变厚而推动现有的SEI层[图5(d)]。进一步地,作者研究了SEI物种的分布,以C2H2-二次离子作为代表性的有机物种,Li2F-作为无机物LiF的代表[图5(e)]。结果显示,从Cu/SEI界面到SEI/电解液界面,Li2F-强度逐渐降低而Li2F-逐渐增加,证实了SEI的双层结构(靠近电解液侧为有机层和靠近电极侧为无机层)。结合SEI生长动力学与其双层结构,可进一步分析得出如下结论:在原始的Cu表面,溶剂分子通过单电子还原为有机物种,电极被有机多孔薄膜覆盖;随着有机层变厚,阴离子和溶剂在多孔有机层难以传输到富含电子的电极表面,因此有机层进一步得电子形成无机物种的SEI内层,如LiF和Li2CO3。这项工作从动力学角度为SEI形成机制提供了直接证据,将有助于为电解液添加剂设计等提供理论指导。
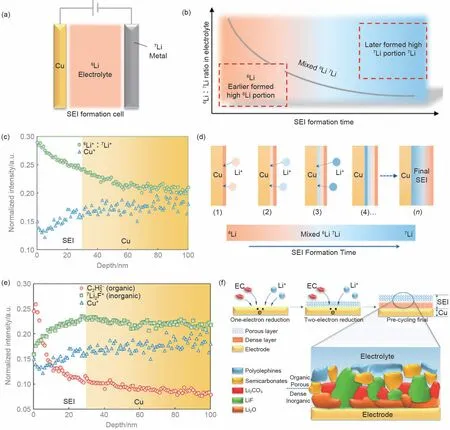
图5 (a)模型电池示意图;(b)SEI形成过程电解液中同位素比率随时间的变化趋势;(c)Cu电极表面SEI的6Li∶7Li比例和Cu+的二次离子SIMS深度分布图像;(d)Cu电极上形成SEI的6Li∶7Li比例随时间变化示意图;(e)Cu电极上C2H2-(有机组分),Li2F-(无机组分)和Cu+二次离子SIMS深度分布图像;(f)SEI形成机制示意图[37]Fig.5 (a)schematic of a model cell;(b)Li isotope ratio variation trend over time in electrolyte during SEI formation process;(c)SIMS depth profiles of the6Li∶7Li ratio and Cu+of SEI on Cu electrode surface;(d)schematic of6Li∶7Li isotope ratio variation over time on SEI of Cu electrode surface;(e)the depth profile of secondary ion of C2H2-(organic),Li2F-(inorganic)and Cu+on Cu electrode;(f)schematic of SEI formation mechanism[37]
2.1.4 离子传输
前几节介绍了SEI的形成和生长过程,本节将介绍Li+如何在SEI中传输。Li+在SEI中的传输一定程度上决定了锂离子电池的性能(尤其是倍率性能)。大部分的研究主要集中在SEI的形成过程、组成和结构等,关于讨论Li+在SEI的传输机制的理解相对较少,其多数为假设或理论计算并无直接的实验证据。
2011年,Lu等[38]利用同位素辅助的ToF-SIMS技术研究了Li+在SEI中的传输机制。图6(a)是SEI处理过程示意图,即首先Cu在1.0mol/L7LiClO4-EC/DEC电解液中放电形成SEI,随后用DMC清洗Cu表面SEI,除去可能吸附的残留电解液并干燥,最后将处理后的SEI浸泡在1.0mol/L6LiBF4-EC/DEC电解液中。图6(b)展示了SEI浸泡于1.0mol/L6LiBF4-EC/DEC电解液后二次离子11B+的深度分布图。无论SEI浸泡时间长短,11B+的深度分布都在外部SEI4~5nm处,表明SEI将6LiBF4限制在SEI的外表面。Li+在SEI的传输可能通过几种不同的机制发生:一种可能是Li+穿过SEI中的孔隙或晶界,由于电中性的要求BF4-随Li+移动。在这种情况下,作为SEI(SEI-Li+)的一部分Li+不受影响。然而,与BF4-不同,如果电解液中Li+与SEI-Li+交换(通过间隙或空位),则电解液中Li+可以穿过SEI而不影响电荷平衡。同位素标记的LiBF4电解质盐,可以区分电解液中Li+和SEI-Li+。在7LiClO4形成的SEI中,6Li+/7Li+比例(自然丰度比值)是恒定的。当SEI浸泡在6LiBF4电解液中,SEI中6Li+/7Li+比例增加则表明电解液中Li+出现在7LiClO4所形成的SEI中。浸泡30s和3min,6Li+/7Li+比例在SEI表面附近达到峰值,在超过5nm的深度后下降到几乎恒定的值。这种趋势与11B+的深度分布相似,表明近SEI/电解液界面的Li+离子的扩散伴随着BF4-的迁移。当浸泡时间为15min时[图6(c)],6Li+/7Li+比例在5nm处达到了峰值并随后逐渐下降,表明电解液/SEI界面上的少量SEI-7Li+会与电解液中6Li+发生交换,可能通过间隙或空位进行传输。因此,在电极SEI多孔外层,电解液中阴离子和阳离子能够共同迁移,在SEI 的无机内层,致密无机物(如Li2O和Li2CO3)则会限制电解质的传输,Li+仅能通过离子交换或空位间隙进行传输。该工作将对SEI层设计的理论设计提供新的思考。
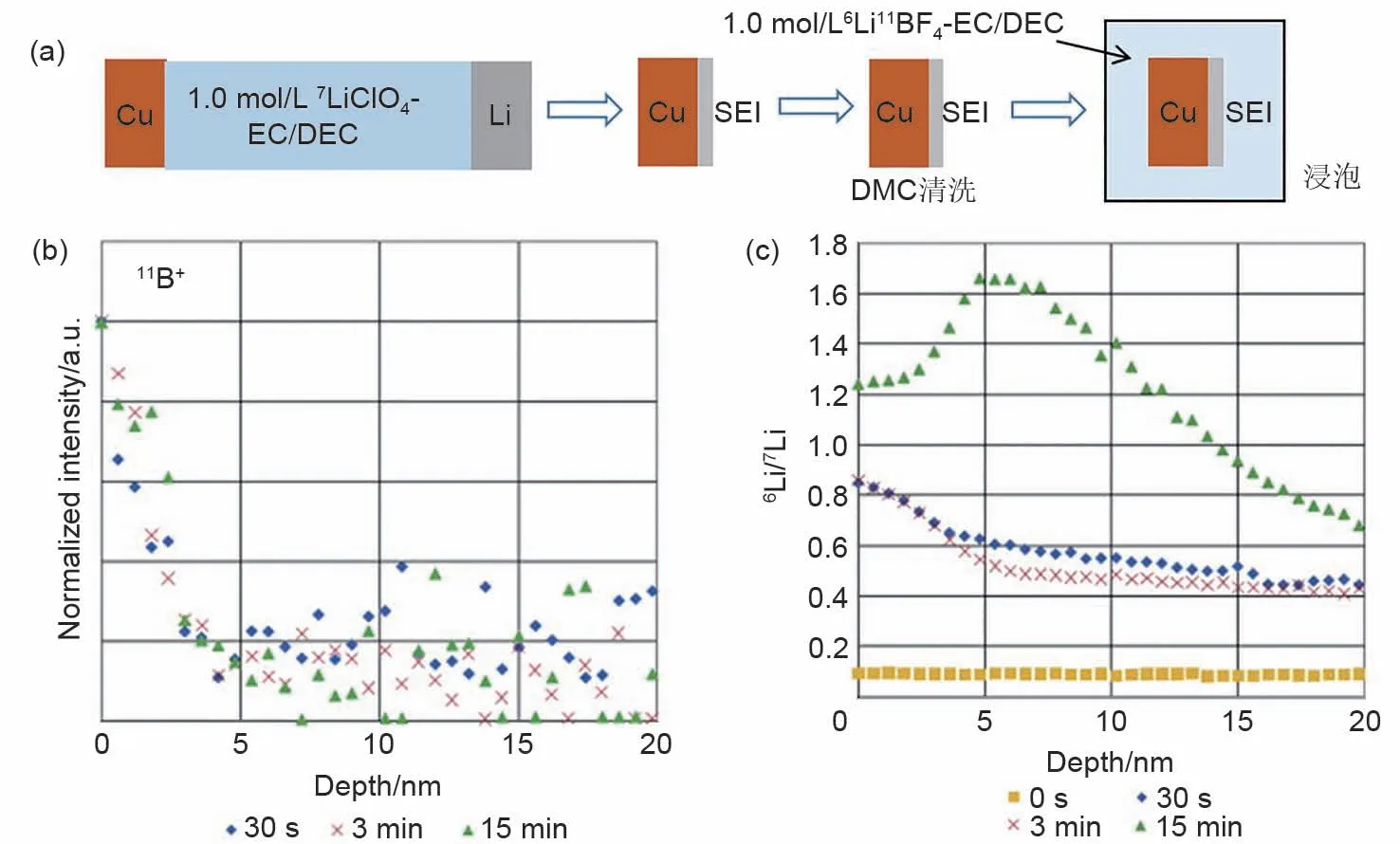
图6 (a)Cu电极表面SEI处理示意图;通过7LiClO4-EC/DEC形成的SEI浸入6LiBF4-EC/DEC电解液后:(b)不同浸入时间的11B+二次离子深度分布;(c)浸入时间3min的11B+和7Li2O+二次离子深度分布[38]Fig.6 Schematic illustration of SEI treatment on Cu electrode surface;SEI formed in7LiClO4-EC/DEC after immersion in6LiBF4-EC/DEC electrolyte:(b)depth profile of secondary ion of11B+at various immersion time,and(c)11B+and7Li2O+at3min immersion time[38]
2.1.5 全固态电池
以上讨论了有机电解液基锂离子电池中的一些反应机制,但是液态电解液存在严重的安全问题(如起火爆炸)。因此,全固态锂离子电池逐渐受到研究者的广泛关注,并被认为是下一代锂离子电池最有希望的候选者。固态电解质的高离子电导率和非可燃性本质有望提高传统锂离子电池的能量密度和安全性[39]。一般来说,全固态锂离子电池正极一般采用固态电解质和活性材料的复合正极,其中大量粒子随机分布并形成复杂的电子和离子导通的三维路径。若活性材料和固态电解质颗粒之间的离子和电子传导路径不畅通,则会导致受限的充放电容量和大的过电势[40]。因此,原位探测电极内部的反应均一性从而优化复合电极结构显得尤为重要。此外,复合电极内部活性材料和固态电解质颗粒的直接物理接触构成电化学界面,在电化学循环过程中由于粒子分布不均产生的局部高电压易导致界面衰退,从而增加界面阻抗[41]。因此,理解电化学界面衰退机制对于改善全固态锂离子电池至关重要。
2021 年,Yamagishi 等[42]利 用 原 位ToF-SIMS技术研究了全固态锂离子电池中锂离子的分布以及电化学界面的衰退过程。图7(a)是原位ToF-SIMS技术测量全固态锂离子电池的示意图,LiNi0.8Co0.15Al0.05O2(NCA)和75Li2S·25P2S5(LPS)为模型研究对象。在电化学测量的同时检测复合电极的横截面,能够原位呈现Li的分布和电化学界面衰退相关的副反应。图7(b)展示了该电池第一圈和第二圈的充放电曲线,可以明显看到放电容量和库仑效率的降低,表明固-固接触界面接触损失以及副反应的发生。如图7(c)所示,在开路状态,6Li+碎片基本均匀分布在整个复合电极上且NCA上的强度低于LPS上的强度。当充电时,6Li+碎片强度降低,表明Li+从NCA颗粒中脱出;放电后,6Li+碎片强度再次略微增加但并没有恢复到开路状态的水平。此外,还观察到NCA中Li2O+碎片(LPS中不含Li2O+),结果发现电池充电后,NCA颗粒上Li2O+碎片的强度明显下降,且再次放电后并不可逆。这些实验现象表明,电子和离子通路局部短缺,非活性NCA粒子较多,Li+难以重新返回NCA粒子中。图7(d)展示了PO2-和PO3-碎片离子强度之和的空间分布演变示意图。PO2-和PO3-碎片离子强度总和改变主要集中在NCA/LPS界面,并且随着循环强度不断增加,表明NCA和LPS之间发生化学/电化学反应产生SEI且不断变厚,因此导致整个电池性能衰退。该工作将有助于研究全固态电池复合电极的优化结构,并提高人们对电池循环过程中降解机制的理解。
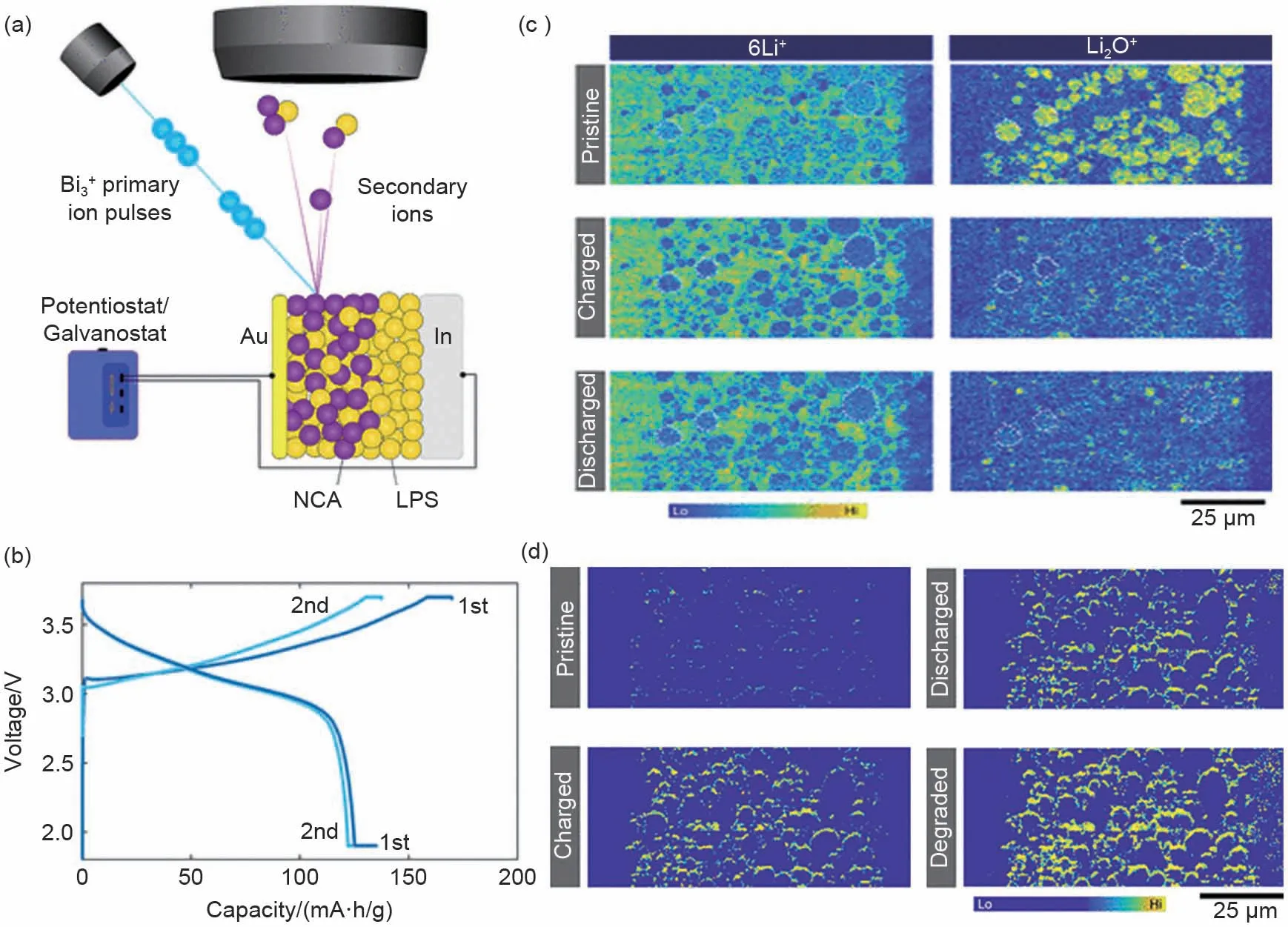
图7 (a)原位ToF-SIMS测量锂离子全固态电池示意图;(b)第一圈和第二圈的充放电曲线;(c)循环期间6Li+和Li2O+碎片的分布演变;(d)PO2-和PO3-二次离子强度在电池循环过程中的演变过程[42]Fig.7 (a)schematic illustration of in situ ToF-SIMS measurement performed on an all-solid-state lithium-ion battery;(b)Charge and discharge profiles for the first and second cycle;(c)evolution of the distribution of6Li+and Li2O+fragments during cycling;(d)evolution of the intensity of PO2-and PO3-secondary ion fragments during the cycling of the all-solid-state lithium-ion battery[42]
2.2锂硫电池
锂硫电池被认为是下一代二次锂电池的最有技术前景的器件之一。大量的工作聚焦于硫正极的改性和设计,并取得了较大进展。但是,高活性的锂金属负极限制了锂硫电池的实际应用。这是因为,金属锂的数量有限,锂离子在沉积和剥离过程的不可逆性严重限制了电池的循环寿命[43-44]。因此,理解金属锂/电解液界面过程对于提高金属锂负极循环寿命至关重要。
2020年,Nanda等[45]使用Ni‖Li2S的无锂负极全电池来研究锂/电解液界面过程,采用ToF-SIMS技术来定量电池循环过程中沉积金属锂的损失。图8(a)比较了LiO-、LiS-和H-二次离子在第5、40和400次循环的深度分布,分别代表Li2O、Li2S以及包含H的界面物种。完全还原的Li2O和Li2S遍及沉积锂的整个厚度,可能嵌入到多孔金属锂基质中。H-二次离子的可能来源于LiH和LiOH。图8(b)比较了LiH-和LiOH-二次离子在第5、40和400次循环时的深度分布,它们的深度分布与H-的深度分布密切相关,因此LiH和LiOH是锂沉积过程的主要含氢组分。此外,C2H3-二次离子的深度分布也遵循与H-相似的趋势,且在整个沉积的锂中具有均匀的信号,醚基电解液的分解产物可能会在沉积的锂中持续分解,形成各种有机组分(含C 和H)。LiO-、LiS-和H-二次离子信号强度在从第5 次到40次循环的过程中并没有明显变化,LiO-的信号强度从第40 次到300 次循环明显增加,表明长期循环过程中沉积的锂中的Li2O量增加。同时,LiS-二次离子的信号强度从第40 到300 个循环也出现了巨大跳跃,因此还原硫物种(Li2S/Li2S2)的数量在多次锂沉积过程中不断积累。相反,H-二次离子以及LiH-、LiOH-和C2H3-二次离子的信号强度从第40次到300 次循环显著降低。因此,在长期循环过程中,含氢的界面成分——LiH、LiOH和含H的有机物种大多在锂沉积过程中耗尽。含氢物种的消耗表明其分解产生气体,如H2、CH4、C2H4、C2H6和H2S。LiOH 和LiH 可能在电池运行期间处于亚稳态,并且可以与残留的水分或电解液反应放出H2。释放出的气体可能会被困在大量沉积的锂中,从而无法通过厚的SEI层逸出。被捕获的气体会通过阻断离子和电子传导通路来严重限制对金属锂的电化学可逆性。如图8(c)和8(d)所示,尽管随着循环圈数增加,锂沉积过程积累了大量的副反应产物,大部分活性锂耗尽后,仍存在大量金属锂。因此,循环过程中,电解液与锂金属的副反应消耗的金属锂实际上并不是导致电池失效的主要原因,而是由于随着循环而不断增厚的SEI 层限制了离子和电子传输,导致“死”锂的产生。此外,多硫物种穿梭到锂负极表面和大量气体的产生也是进一步限制金属锂可逆沉积的可能性原因。该工作定量评估了金属锂在长期循环过程中的变化,对于改善锂硫电池循环寿命具有指导性的意义。
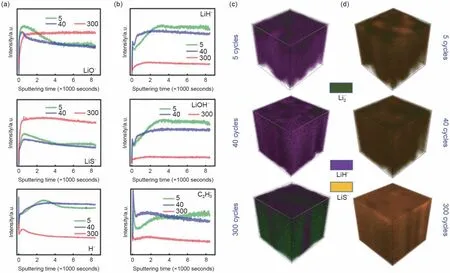
图8 (a)锂负极表面LiO-、LiS-和H-二次离子在第5、40以及300圈循环的深度分布,分别代表Li2O、Li2S以及包含H的界面物种;(b)锂负极表面LiH-、LiOH-和C2H3-二次离子在第5,40以及300圈循环的深度分布,分别代表LiH、LiOH以及各种有机界面物种;(c)、(d)在第5,40以及300圈循环后Li 2-、LiH-和LiS-的SIMS扫描3D重建图像[45]Fig.8 (a)depth profiles of LiO-,LiS-and H-secondary ions that represents respectively Li2O,Li2S and various hydrogen-containing interphasial species,at 5,40 and 300 cycles on lithium anode;(b)depth profiles of LiH-,LiOH-and C2H3-secondary ions that represents respectively LiH,LiOH and various organic interphasial species,at 5,40,and 300 cycles on lithium anode;(c),(d)3D reconstructions of the SIMS signal for LiH-and LiS-at 5,40,and 300 cycles[45]
2.3 锂氧电池
非水溶剂锂氧电池由于超高的理论能量密度而备受研究者关注。但是,实际的锂氧电池仍然面临诸多挑战,如循环寿命低、倍率能力差、过电势高等[46]。深入理解锂氧电池充放电反应原理,揭示其性能受限因素至关重要。研究发现,锂氧电池放电(即氧还原反应)形成产物过氧化锂(Li2O2)易导致电极堵塞,通常在一段恒定电压的放电平台后,电压急剧下降导致其放电容量远远低于理论预期。此外,锂氧电池充电电势也取决于放电过程的速率和深度[47]。因此,理解电极界面Li2O2形成和分解的反应位点和传输限制对于改善锂氧电池充放电性能显得至关重要。
2019年,Wang等[48]选择了两种模型电极,即碳电极和碳负载催化剂Ru 的碳电极,以揭示放电过程氧反应界面。将锂氧电池首先在16O2气氛中放电至1000 mA·h/g,随后在18O2气氛中放电至2000 mA·h/g,构建了特殊设计的放电产物Li2O2结构。为了揭示其化学结构,以二次离子为18O-指示剂,利用ToF-SIMS获得放电产物的三维化学信息。图9 显示了两种模型电极的ToF-SIMS 深度扫描18O-的3D分布图像。随着溅射深度增加,18O-的信号强度逐渐降低,当溅射至距电极表面175 nm时,两个电极的18O-信号几乎消失。18O-信号分布表明后者形成的Li218O2位于放电产物的顶面。进一步地,使用微分电化学质谱研究了锂氧电池充电过程,发现充电过程首先释放18O2,随后是16O2。因此,无论有无催化剂的锂氧电池充放电反应界面均位于Li2O2/电解液界面。该研究为可充电锂氧电池的反应机制提供重要见解,开发的同位素标记的ToF-SIMS共性技术可普遍应用于揭示其他金属-氧气(Na/K-O2)电池的反应机制。
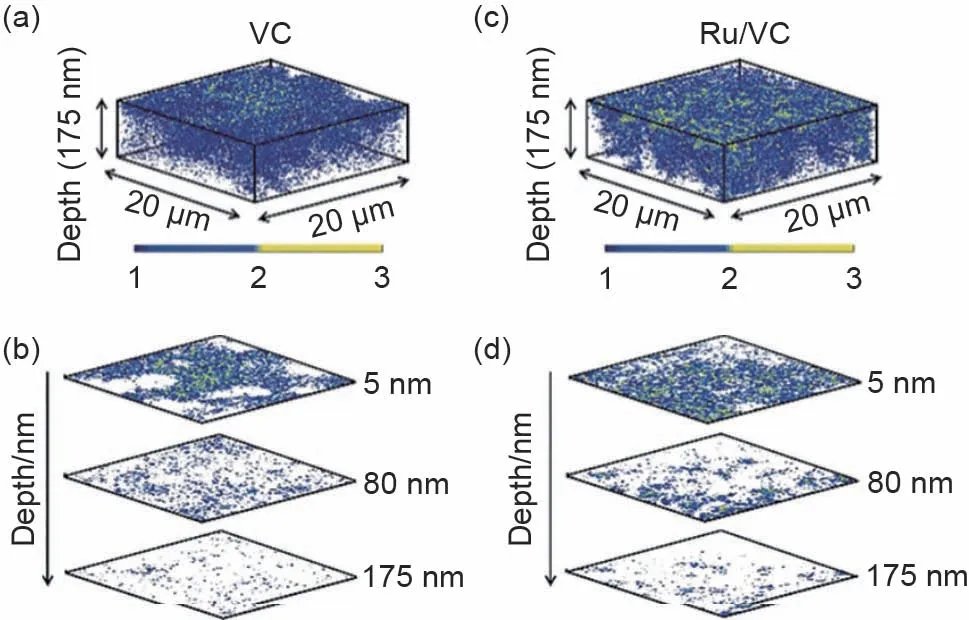
图9 放电电极上ToF-SIMS深度扫描二次离子18O-的3D图像:(a)18O-在碳(VC)电极上的3D分布;(b)VC电极上18O-重建的3D图像中选择的三个厚度层;(c)催化剂负载的碳电极(Ru/VC)电极上18O-的3D分布;(d)Ru/VC电极上重建3D图像中选择的三个厚度层[48]Fig.9 3D distribution of secondary ion18O-on the discharge electrode with ToF-SIMS depth scan:(a)3D distribution of18O-on the carbon(VC)electrode;(b)selected layers from the reconstructed 3D image of18O-on the VC electrode at three depths;(c)3D distribution of18O-on the carbon load with Ru(Ru/VC)electrode;(d)selected layers from the reconstructed 3D image of18O-on the Ru/VC electrode at three depths[48]
3 总结和展望
高效和安全的电化学储能器件是目前研究者和消费者普遍关心的问题。随着社会经济发展和能源消费的不断升级,电化学储能科学事业必将迎来快速发展。为此,必须对电化学储能器件进行基础研究,提高对电化学反应的深入理解,特别是电极/电解液界面反应。为此,本文总结了最近ToFSIMS 技术(包括非原位和电化学联用的原位技术)在能源电化学中的应用和进展。ToF-SIMS 技术是一种研究界面电化学反应的强有力工具,已经被广泛应用于能源电化学的基础研究。ToF-SIMS 具有低检测限和分辨率高等优点,能够全方位收集电极表面分子/原子信息。重要的创新是微流控技术将电化学和ToF-SIMS 技术耦合到一起,允许实现电化学不稳定反应中间体的检测以及电极/电解液界面过程的可视化。这些基础见解必将厘清电化学界面反应机制(如反应路径、反应动力学等),理解储能器件劣化的根本原因,从而理性设计电化学储能材料并最终制备高效安全的电化学储能器件。然而,原位电化学ToF-SIMS 联用技术的应用仍然具有一定的限制条件,如涉及气体的电化学反应。而且,原位电化学ToF-SIMS 的使用仅限于少数专门的实验室,该技术需要进一步的创新以实现该技术的全面推广。ToF-SIMS技术(尤其是原位电化学联用技术)的未来发展应该进一步缩短时间和提高空间分辨率,实现物理化学信息的纳米级甚至亚纳米级和超快时间分辨,为能源电化学界面过程提供新的见解从而进一步改善储能器件性能。除此之外,ToF-SIMS 技术与其他先进谱学联用技术预计可以提供完整的电化学界面物理化学图像,增加对电化学反应机制的全面理解(反应路径、反应位、反应动力学等)。总之,电化学ToF-SIMS联用技术已经取得了重要进步,随着仪器方法的进一步创新(如环境质谱电离方法的发展),相信电化学质谱联用技术将能够实现储能器件工况条件下的原位检测,电化学质谱研究方法也将在现代和未来能源电化学中扮演不可或缺的重要角色。
