CoOx用于负载Pd催化剂的CO和C3H8氧化
2022-03-18钟栎锭吴玉瑾
钟栎锭,吴玉瑾,王 丽,郭 耘
(华东理工大学 化学与分子工程学院,工业催化研究所,上海 200237)
一氧化碳(CO)和丙烷(C3H8)作为主要的大气污染物,大部分来源于化石燃料未充分燃烧的工业废气或机动车尾气,威胁着环境和人类健康[1-2]。催化氧化可以有效实现CO和C3H8的净化。与非贵金属氧化物催化剂相比,负载贵金属催化剂具有反应温度低、环境适应性好等特点[3-4]。负载Pd催化剂是当前研究较为广泛的贵金属催化剂,其催化性能与Pd的分散程度、化学状态及载体种类密切相关[5]。Xu等[6]通过密度泛函理论计算(DFT)得到单原子Pd具有良好的氧化活性和稳定性。Pd的化学状态与氧化反应性能紧密相关,并受到反应物体系的影响。据报道,金属态Pd0和非化学计量比PdOx<1有利于CO氧化[7];而Zhang等[8]则认为Pd+比PdO物种更有利于CO吸附和活化。与CO氧化相反,氧化态Pd比金属Pd具有更高的C3H8氧化活性[9-11],Wang等[11]发现Pd2+可以促进催化剂的C3H8氧化。
载体的氧化性能影响载体与贵金属之间的相互作用,可还原性载体与贵金属之间的相互作用较强,而惰性载体与贵金属之间的相互作用相对较弱[12]。载体与贵金属之间的强相互作用会影响贵金属的化学状态。例如,Pd与立方体CeO2之间的强相互作用使得Pd以PdxCe1-xO2-σ固溶体形式存在,但Pd与八面体CeO2之间的弱相互作用使得Pd以PdOx颗粒形式存在[13]。此外,载体与贵金属之间的强相互作用还会影响载体-贵金属界面的原子数[14]。Jin等[15]研究发现,当Pd纳米颗粒的尺寸从18 nm减小到6 nm时,催化剂在140 ℃条件下的转化率(TOF值)提高了10倍。
对于负载Pd催化剂而言,O2的活化对氧化性能至关重要,引入具有氧化性能的活性中心可以进一步提升催化剂的氧化性能[16]。Co3O4因具有较多的表面缺陷、丰富的表面吸附氧物种及优异的耐水和耐硫性能[17],被广泛用于催化氧化反应[18-19]。考虑到载体的氧化还原性能会影响贵金属的化学状态,而Co3O4的引入能够促进氧活化,本文以CeO2和Al2O3为载体,采用沉积沉淀法制备负载Pd催化剂,并分别考察CoOx引入对负载Pd催化剂CO和C3H8氧化性能的影响。在此基础上,利用N2吸脱附、X射线衍射(XRD)、氢气程序升温还原(H2-TPR)、X射线光电子能谱(XPS)、拉曼光谱(Raman)等技术对催化剂特性进行表征,并采用原位红外光谱(insituDRIFTS)探讨CoOx引入对催化剂氧化反应性能产生影响的作用机制。
1 材料与方法
1.1 材料与试剂
实验所用的化学药品均为分析级。硝酸钯(Pd(NO3)2)购自国药集团试剂有限公司,硝酸钴(Co(NO3)2·6H2O)购自北京伊诺凯科技有限公司,氢氧化钠(NaOH)购自上海阿拉丁生化有限公司,无水乙醇购自上海泰坦科技股份有限公司。
1.2 Pd/CoOx/Al2O3和Pd/CoOx/CeO2的制备
Pd/CoOx/Al2O3的制备:将Co(NO3)2·6H2O和Al2O3溶解于去离子水中,与配好的一定浓度的NaOH混合,经80 ℃搅拌、100 ℃干燥和550 ℃焙烧后,得到CoOx/Al2O3纳米颗粒,标记为CoOx/Al2O3。将Pd(NO3)2溶液逐滴加入到CoOx/Al2O3粉末中,超声使液体分散均匀。所得样品先在室温放置24 h,再经80 ℃干燥和550 ℃焙烧4 h,得到Pd/CoOx/Al2O3催化剂。
Pd/CoOx/CeO2的制备:使用CeO2替代Al2O3后,重复上述步骤,得到Pd/CoOx/CeO2催化剂。
1.3 催化剂的表征
比表面积测定在NOVA 4200e比表面和孔隙度分析仪上进行。测试前所有样品均在180 ℃条件下真空脱附6 h,比表面积采用BET(Brunauer-Emmett-Teller)方法进行计算。
等离子体电感耦合发射光谱(ICP-AES)表征在TJAIRIS 1000型等离子体电感耦合发射光谱仪上进行,用于检测不同元素含量。
XRD表征在Bruker D8 Focus型X射线衍射仪上进行。入射光源为Cu靶Kα 射线(λ=0.154 06 nm),管电压为40 kV,管电流为40 mA,扫描速率为6(°)/min,扫描范围为10°~80°。
H2-TPR表征在AutoChem Ⅱ全自动化学吸附仪上进行。称取50~80 mg催化剂样品,通入体积分数为10%的H2/N2混合气(40 mL/min),待检测信号稳定30 min后,以10 ℃/min的速率升温至600 ℃。通过TCD检测器记录信号值,并以商业CuO还原为参比,对样品还原过程中的H2消耗进行定量。
Raman光谱测试在inVia Reflex显微拉曼光谱仪上进行。激光发射源为Ar+离子激光(514.5 nm),光斑约为1 nm,入射光功率约为2 mW,精确度约为3.5 cm-1。
XPS表征在KratosAXIS UltraDLD型X射线光电子能谱仪上进行。激发光源采用Al靶Kα射线(1 486.6 eV),以C 1s(284.8 eV)为内标对样品进行结合能矫正。
insituDRIFTS表征在Nicolet Nexus 670光谱仪上进行。检测器为MCT,样品池窗口为KBr。检测器用液氮冷却,采用4 cm-1分辨率,扫描64次。实验过程如下:样品研磨后放入样品池,先用体积分数10%的H2/Ar气体(20 mL/min)在200 ℃条件下预处理1 h,再切换为氩气气氛。CO氧化反应在50 ℃条件下进行,先通入体积分数1%的CO/Ar气体(50 mL/min)至吸附饱和,再通入体积分数20%的O2/Ar气体(50 mL/min)至吸附饱和。C3H8氧化反应在200 ℃条件下进行,先通入体积分数1%的C3H8/Ar气体吸附30 min,再通入体积分数1%的C3H8和10%的O2吸附30 min,最后用Ar吹扫至饱和。
1.4 活性测试
催化剂的氧化活性评价在固定床反应器上进行。CO氧化反应的催化剂用量为50 mg,反应原料气为1% CO+20% O2+79% Ar(体积分数),气体总流量为50 mL/min。C3H8氧化反应的催化剂用量为100 mg,反应原料气为0.2% C3H8+2% O2+97.8% Ar(体积分数),气体总流量为50 mL/min。评价前催化剂在200 ℃条件下还原预处理。反应物和产物分析由色谱(GC 2060)完成,采用FID检测器并配备甲烷转化炉,填充柱为PT柱(3 m)。CO或C3H8转化率由反应前后CO或C3H8的浓度变化计算得到。在排除内扩散和外扩散,并控制转化率低于15%的前提下,进行动力学测试。CO氧化反应温度为50 ℃,C3H8氧化反应温度为200 ℃。不同温度下的反应速率通过阿伦尼乌斯公式计算活化能。
2 结果与讨论
2.1 CoOx引入对CO和C3H8氧化性能的影响
图1为负载Pd系列催化剂的CO催化氧化活性曲线。Pd/CeO2和Pd/Al2O3可分别在65 ℃和125 ℃实现CO的全转化,表明Pd/CeO2对CO的氧化性能高于Pd/Al2O3。CoOx引入可以提高Pd/CeO2和Pd/Al2O3的CO催化氧化活性,使反应的全转化温度向低温偏移。CeO2载体催化剂的CO催化氧化性能高于Al2O3载体催化剂,其中Pd/CoOx/CeO2比Pd/Al2O3的全转化温度低80 ℃。

图1 负载Pd系列催化剂的CO催化氧化活性Fig.1 Catalytic activities of the Pd supported catalysts for CO oxidation
由负载Pd系列催化剂的C3H8催化氧化活性曲线(图2)可知,CoOx的引入也可以提升Pd/Al2O3和Pd/CeO2的C3H8催化氧化活性,使二者的全转化温度分别向低温移动30 ℃和85 ℃。虽然Pd/CoOx/CeO2对C3H8的完全氧化温度较Pd/CeO2降低85 ℃,但其完全氧化温度仍低于Pd/CoOx/Al2O3(330 ℃)。
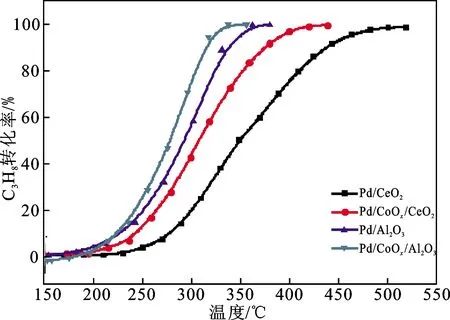
图2 负载Pd系列催化剂的C3H8催化氧化活性Fig.2 Catalytic activities of the Pd supported catalysts for C3H8 oxidation
在此基础上,进行负载Pd系列催化剂的动力学测试,获得动力学反应参数,结果列于表1。反应速率结果进一步表明,CoOx引入对氧化反应的促进作用与反应体系、反应物密切相关。CoOx引入显著提升了Pd/CeO2的氧化性能,使其CO氧化反应速率提高1.8倍,C3H8氧化反应速率提高2.7倍。同样,CoOx引入也可以显著增强Pd/Al2O3的氧化性能,使其CO氧化反应速率提高2倍,C3H8氧化反应速率提高1.5倍。

表1 负载Pd系列催化剂的氧化反应速率、结构和组成Tab.1 The reaction rates, texture structures and compositions of the Pd supported catalysts
2.2 XRD和比表面积表征
如表1所示,各催化剂的Pd和CoOx实际负载量差异不大,Pd质量分数在0.82%左右,Co质量分数在0.45%左右。N2吸脱附实验结果表明,CoOx引入对催化剂的比表面积影响不显著。
图3为制备催化剂的XRD谱图。由图3a可知,CeO2为典型的氧化铈立方萤石结构(JCPDS 34-0394); 而Pd和CoOx引入并未观察到新的特征衍射峰。Pd和CoOx引入后,CeO2的特征衍射峰向高角度偏移,这是由于Pd和CoOx进入CeO2晶格形成了固溶体[20]。Pd/CeO2和Pd/CoOx/CeO2的晶粒尺寸均在6 nm以下(表1)。由图3b可知,载体氧化铝为γ-Al2O3;负载CoOx和Pd后,Al2O3的XRD谱图中也没有检测到新的衍射峰,这可能与CoOx和Pd在催化剂表面高度分散或者低于XRD检测限相关。

图3 催化剂的XRD谱图Fig.3 XRD patterns of the catalysts
2.3 H2-TPR表征
催化剂的H2-TPR谱图如图4所示,相应的耗氢量结果列于表2中。CeO2在300~600 ℃高温区出现有1个宽化的还原峰,归属于CeO2表面晶格氧的还原[21-22],其耗氢量为518 μmol/g。引入CoOx后,Co与Ce之间的相互作用使该还原峰低温偏移至200~500 ℃,其耗氢量为648 μmol/g。虽然通过分峰处理可获得CoOx和CeO2的耗氢量,但由CoOx的理论耗氢量60 μmol/g (Co3+→Co2+)可知,CoOx引入对CeO2还原的促进作用不明显。负载Pd后,Pd/CoOx/CeO2和Pd/CeO2分别在170 ℃和130 ℃处出现有1个还原峰,归属于PdO的还原[23];对应的耗氢量分别为1 757 μmol/g和1 433 μmol/g,远大于PdO到Pd0的理论耗氢量(82 μmol/g),结合CoOx的理论耗氢量可知,Pd的引入有效促进了CeO2还原。

图4 催化剂的H2-TPR谱图Fig.4 H2-TPR spectra of the catalysts
Pd/Al2O3在66 ℃出现有明显的归属于Pd物种的还原峰,其耗氢量(57 μmol/g)略低于PdO到Pd0的理论耗氢量(82 μmol/g),而Pd/CoOx/Al2O3在87 ℃的还原峰耗氢量(117 μmol/g)仍然低于 PdO和CoOx的理论耗氢量之和,表明催化剂表面含有部分金属态Pd。

表2 催化剂的H2-TPR、XPS和Raman数据Tab. 2 H2-TPR,XPS and Raman data of the catalysts
2.4 Raman表征
图5a为CeO2系列催化剂的Raman谱图。CeO2载体的Raman谱图中有4个振动峰,分别位于260、461、598 和1 174 cm-1处。位于461 cm-1处的强峰为CeO2面心立方晶格的F2g拉曼振动峰(D-mode),由Ce4+周围氧的对称伸缩振动产生;位于1 174、598和260 cm-1处的3个弱峰分别为二阶纵向振动峰(2LO)、CeO2立方萤石结构的缺陷诱导模式峰(D)和二阶横向振动峰(2TA-mode)[24]。
I(1 174+598)/I(461)峰强度比值反映了样品表面的缺陷程度[25]。CeO2载体的I(1 174+598)/I(461)比值仅为0.067,Pd的引入使I(1 174+598)/I(461)比值增加到0.221,而CoOx的引入使I(1 174+598)/I(461)比值进一步增加到0.411,表明Pd和CoOx的引入能够增加CeO2表面氧缺陷(表2)。
图5b为Al2O3系列催化剂的Raman谱图。Pd/Al2O3和Pd/CoOx/Al2O3在639 cm-1处出现有振动峰,归属为PdO的振动峰[26]。

图5 催化剂的Raman谱图Fig.5 Raman spectra of the catalysts
2.5 XPS表征
图6为催化剂的Pd 3d、O 1s、Ce 3d和Co 2p XPS谱图,相应的计算结果列于表2。图6a为催化剂的Pd 3d XPS谱图。结合能为335.4、337.1和337.9 eV的峰分别归属于金属态Pd0、PdOx纳米颗粒中的Pd2+及表面或次表面PdxCe1-xO2-σ固溶体中的Pdx+[27]。积分处理可知,Pd/CoOx/CeO2表面Pd主要以PdxCe1-xO2-σ固溶体形式存在,而Pd/Al2O3表面Pd主要以金属态Pd0形式存在。这是因为Pd与载体Al基之间的相互作用较弱,该结果与H2-TPR表征结果一致。
图6b为催化剂的O 1s XPS谱图。Pd/CoOx/CeO2和Pd/CeO2表面有2种氧物种,而Pd/Al2O3和Pd/CoOx/Al2O3表面仅有1种氧物种。结合能为529.5 eV的峰归属于表面晶格氧(Oα),结合能为531.1~531.5 eV的峰归属于表面吸附氧(Oβ)。Oβ/Oα摩尔比可用来表征氧空位浓度,Pd/CoOx/CeO2的Oβ/Oα摩尔比比Pd/CeO2高0.083 7(表2),说明CoOx的加入增加了催化剂的表面氧物种。
图6c为催化剂的Ce 3d XPS谱图。与Pd/CeO2相比,Pd/CoOx/CeO2表面Ce3+离子的比例增加(表2)。图6d为催化剂的Co 2p XPS谱图。Co3+和Co2+XPS峰的结合能分别为780.3 eV和781.9 eV,位于786.3 eV的卫星峰对应于Co2+物种[28]。Pd/CoOx/CeO2的Co3+/Co2+摩尔比(0.640 2)高于Pd/CoOx/Al2O3(0.205 3,表2),而Co3+是催化剂CO氧化的活性中心,具有高含量Co3+的Pb/CoOx/CeO2更有利于CO氧化[29]。
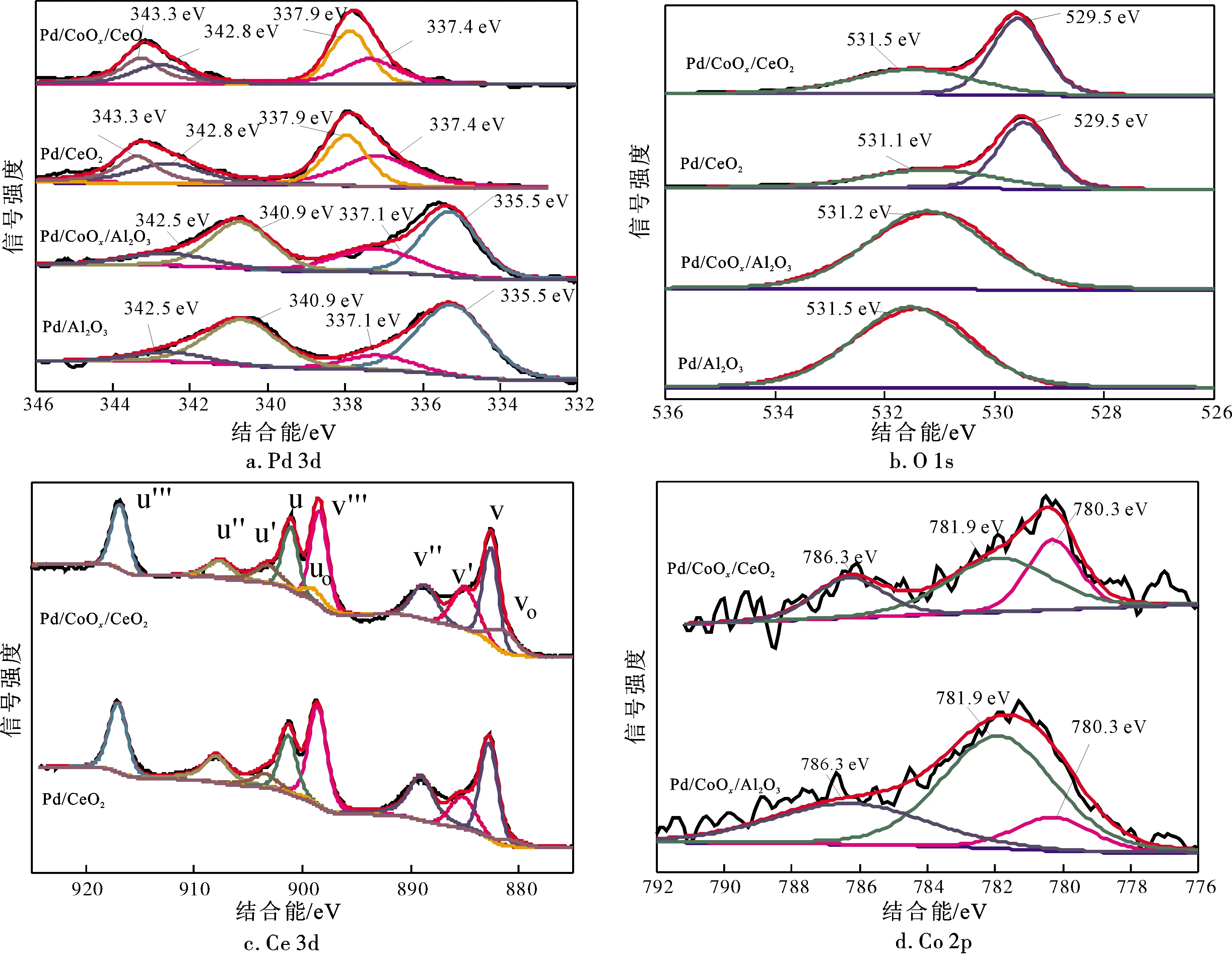
图6 催化剂的XPS谱图Fig.6 XPS spectra of the catalysts
2.6 in situ DRIFTS分析
图7a、7b为CeO2系列催化剂在50 ℃条件下的CO吸附、氧化和脱附原位漫反射红外谱图。位于2 300~2 400 cm-1的振动峰归属于气相CO2,位于1 000~1 800 cm-1的振动峰归属于碳酸盐,2 090、2 117和2 173 cm-1处的振动峰分别归属于Pd0-CO线性吸附、Pd+-CO线式吸附和Pd2+-CO吸附。
如图7a所示,Pd/CeO2在通入CO 2 min时,表面检测到Pd0-CO振动峰(2 090 cm-1)、Pd2+-CO振动峰(2 173 cm-1)、双齿碳酸盐振动峰(1 025、1 267 cm-1)、单齿碳酸盐振动峰(1 387、1 470、1 583 cm-1)[30-31],说明CO可以与Pd/CeO2表面吸附的氧反应,且产物以碳酸盐为主。将反应气切换为O26 min后,Pd0-CO振动峰(2 090 cm-1)消失,而碳酸盐振动峰强度没有增加,说明Pd0-CO氧化后生成的产物不是碳酸盐物种。
如图7b所示,Pd/CoOx/CeO2在通入CO 2 min时,表面检测到Pd0-CO(2 080 cm-1)、双齿碳酸盐(1 290 cm-1)、单齿碳酸盐(1 401、1 583 cm-1)和气相CO2(2 300~2 400 cm-1)的振动峰。与Pd/CeO2相比,Pd/CoOx/CeO2的Pd0-CO振动峰向低波数方向偏移,表明Pd与Co之间存在电子转移,影响了对CO的吸附。此外,CO2的生成表明CO氧化后的产物除了碳酸盐,还有一部分CO2。将反应气切换成O2后,Pd/CoOx/CeO2在2 117 cm-1处出现了1个归属于Pd+-CO的新振动峰,表明O2的加入能将部分Pd0氧化为Pd+。通入O28 min后,CO的振动峰消失,但依然能检测到CO2的振动峰,表明Pd+具有更高的氧化性能。

对以CeO2为载体的催化剂而言,CoOx引入能够促进金属态Pd物种氧化,形成具有更高活性的Pd+物种,同时抑制催化剂表面碳酸盐物种的生成。

图7 50 ℃条件下CO吸附、氧化和脱附的原位红外谱图Fig. 7 In situ DRIFTS spectra of adsorption,oxidation and desorption of CO at 50 ℃
图8为催化剂在200 ℃条件下C3H8吸附、氧化和脱附过程的原位漫反射红外谱图。2 800~3 000 cm-1的吸收峰归属于气相丙烷振动峰、1 288 cm-1的吸收峰归属于C—H变形振动峰、1 623 cm-1的吸收峰归属于C=C伸缩振动峰、3 500 cm-1的吸收峰归属于Ce-OH振动峰[32],1 419、1 561、1 730 cm-1的吸收峰归属于羧酸盐振动峰。
如图8a所示,Pd/CeO2在通入C3H830 min时,未出现明显的振动峰,表明C3H8在Pd/CeO2表面的吸附能力较弱。当通入C3H8+O2混合气 4 min时,Pd/CeO2在1 419、1 561、1 730 cm-1处出现了较强的羧酸盐振动峰,使用Ar吹扫后羧酸盐依然存在,说明其在Pd/CeO2表面较为稳定。
如图8b所示,Pd/CoOx/CeO2在通入C3H830 min时,表面出现νas(COO)振动峰(1 541 cm-1)以及生成的气相CO2振动峰(2 300~2 400 cm-1),表明CoOx的引入可以促进C3H8的吸附并将其氧化。通入C3H8+O2混合气4 min时,Pd/CoOx/CeO2出现羧酸盐振动峰(1 419 cm-1)、气相C3H8振动峰(2 966、2 931 cm-1)和气相CO2振动峰(2 300~2 400 cm-1)。说明C3H8氧化会导致羧酸盐在表面积累。使用Ar吹扫后,2 966、2 931和1 541 cm-1处的振动峰消失,而CO2振动峰强度增加,表明催化剂表面部分羧酸盐可转化生成CO2。由此可知,CoOx的引入能够促进C3H8吸附,使其快速氧化;此外,也有利于氧化中间产物羧酸盐的分解。

图8 200 ℃条件下C3H8吸附、氧化和脱附的原位红外谱图Fig.8 In situ DRIFTS spectra of adsorption, oxidation and desorption of C3H8 at 200 ℃
如图8c所示,Pd/Al2O3在通入C3H830 min时,在1 623 cm-1处出现C=C伸缩振动峰。当通入C3H8+O2混合气 4 min时,Pd/Al2O3在2 865、2 965 cm-1处出现弱的气相C3H8振动峰。Ar吹扫后发现C3H8在Al2O3表面的吸附能力较弱。
如图8d所示,Pd/CoOx/Al2O3在通入C3H830 min时,除观察到C=C伸缩振动峰(1 623 cm-1)外,还出现有羧酸盐振动峰(1 561 cm-1)、气相C3H8振动峰(2 965 cm-1)和CO2振动峰(2 300~2 400 cm-1),羧酸盐的出现表明CoOx的引入促进了催化剂表面C3H8的吸附和氧化。在通入C3H8+O2混合气后,气相CO2振动峰(2 300~2 400 cm-1)进一步增强,但未发现表面羧酸盐的变化。使用Ar吹扫后,C3H8振动峰和羧酸盐振动峰消失,而CO2振动峰增强,说明羧酸盐物种可转化为CO2,并从催化剂表面脱附。
催化剂对反应物的吸附以及对O2的吸附和活化都会影响其对CO和C3H8氧化的反应性能。对于CO氧化而言,负载Pd催化剂对CO的吸附能力较强,从而弱化了对O2的吸附和活化,不利于反应性能的提高。为了解决竞争吸附的问题,Qiao等提出了“双活性中心”的概念,即通过Pd实现对CO的吸附和活化,由可还原载体Fe(OH)x作为活化氧中心,其中可还原的金属氧化物成为氧的主要来源[33]。Co3O4对O2具有良好的吸附和活化能力,同时其表面晶格氧也可直接参与氧化反应,因此Co的引入可以解决CO吸附与O2活化之间的竞争矛盾。H2-TPR结果发现:Pd与CoOx之间的相互作用促进了CoOx在较低温度下的还原(Pd/CoOx/Al2O3),进而增加了催化剂表面在低温下的活性氧物种。在CO吸附的FTIR实验中,Pd/CoOx/Al2O3生成的CO2高于Pd/Al2O3,也进一步证实了上述结论。此外,计算H2-TPR中耗氢量发现:Pd、CoOx、可还原载体三者之间的相互作用可进一步促进载体(CeO2)还原,增加Pd/CoOx/CeO2表面可还原物种的数量。CoOx的引入可提高催化剂表面可还原氧的数量,从而提高氧化反应的性能。
贵金属Pd的存在状态受载体氧化还原性的影响。在可还原载体上,Pd以氧化态形式存在,如PdO2或者PdxCe1-xO2-σ固溶体中的Pdx+;而在惰性载体上,Pd以金属态Pd0和PdO2的形式存在。CoOx的引入也会影响催化剂表面Pd的化学状态。CoOx、Pd、CeO2之间相互作用的增加使得表面Pd主要以Pdx+的形式存在;而Pd与CoOx的相互作用也会增加氧化Pd物种。
结合原位红外结果发现:对CO氧化催化剂而言,CoOx的引入能够促进Pd/CeO2中的Pd物种被还原为高活性的Pd+物种,并抑制催化剂表面碳酸盐物种的生成;而在Pd/Al2O3体系中,CoOx可以作为活性氧中心,避免了CO和O2之间的竞争吸附对反应的抑制作用。对C3H8氧化催化剂而言,CoOx引入促进了C3H8在Pd/CoOx/CeO2表面的吸附,其C3H8氧化产物分别为羧酸盐和CO2,随着氧化反应的进行,稳定羧酸盐生成和积累。相反,C3H8在Pd/Al2O3上的吸附能力相对较强,所以CoOx引入促进了氧化中间产物羧酸盐的分解。
3 结论
通过沉积沉淀制备出 Pd/CeO2和Pd/Al2O3催化剂,并考察了CoOx引入对CO和C3H8氧化的影响。CoOx的引入有效促进了Pd/CeO2对CO氧化、Pd/Al2O3对C3H8氧化性能的提高。CoOx对反应的促进作用与载体的氧化还原性和反应物密切相关。CoOx引入可增加Pd与惰性载体之间以及CoOx、 Pd、可还原载体三者之间的相互作用,提高催化剂表面活性氧的数量,增加氧化态Pd物种的数量。在CO氧化反应中,引入CoOx能够促进Pd/CeO2表面Pd物种在反应气氛下生成具有高活性的Pd+物种,抑制碳酸盐物种生成;Pd/Al2O3中CoOx对氧的活化作用可以避免CO和O2的竞争吸附,提高CO氧化反应性能。在C3H8氧化反应中,CoOx的引入可以促进C3H8在Pd/CoOx/CeO2表面的吸附,其C3H8氧化产物分别为羧酸盐和CO2,氧化反应的进行会导致稳定羧酸盐的生成和积累;Pd/Al2O3中CoOx的引入能够有效促进氧化中间产物羧酸盐的分解,提高催化剂的反应性能。
