直接进样-超高效液相色谱-串联质谱法同时测定水中14种有机污染物
2022-03-16叶燕慧刘阳春王东霞赵红磊
叶燕慧,刘阳春,王东霞,赵红磊,郭 伟
(北京市水文总站,北京 100089)
水中有机污染物种类繁多,危害性不同,其中丙烯酰胺、灭草松、2,4-滴、乐果、苦味酸、联苯胺、五氯酚、呋喃丹、甲萘威、阿特拉津、内吸磷、马拉硫磷、对硫磷和毒死蜱为水体中常见的14种有机污染物。联苯胺具有强致癌性,被国际癌症研究机构(IARC)列为第一类致癌物[1];丙烯酰胺被IARC列为第2类致癌物;五氯酚、乐果、对硫磷是我国水中优先控制的污染物[2];苦味酸为高毒类物质;以呋喃丹、甲萘威、毒死蜱、马拉硫磷、内吸磷为代表的杀虫剂和以阿特拉津、灭草松、2,4-滴等为代表的除草剂是我国使用较多的农药。上述14种有机污染物对人体危害性较强,均属于我国现行国标严控指标项目[3-5],并配套了相应的检测方法,规定了最高限值。标准中指定的测试方法需对大体积水样富集处理后方可进行分析,前处理过程冗长,试剂用量大,易对实验人员的身体健康造成危害。目前,固相萃取法已广泛用于有机物的分离富集,虽然较液-液萃取可节省大量溶剂,提高分析效率,但仍需大量水样富集,操作繁琐,有时甚至可产生假阳性结果。液相色谱-质谱是一种灵敏度高,选择性好,分析速度快的定性、定量分析方法。液相色谱-串联质谱(LC-MS/MS)法已逐渐应用于水中痕量有机污染物的测定[6-8],其中直接进样法也得到了一定的应用[9-13]。如,张明等[13]建立了直接进样-超高效液相色谱-串联质谱(UPLC-MS/MS)法快速测定地表水中联苯胺、苦味酸、甲萘威、阿特拉津和溴氰菊酯,该方法简便快捷、灵敏准确。目前,已逐步开展水中部分理化性质相近化合物的多组分同时检测研究,但不同理化性质化合物的多组分同时检测的分析方法还较缺乏。此外,不同极性的目标化合物在色谱柱上的吸、脱附效率不同,在同时进样检测时通常存在相互干扰的现象。
高效液相色谱-串联质谱法测定有机物大多采用多反应监测模式,为保证每种化合物的检测灵敏度,需分时段监测。动态多反应监测(DMRM)可根据色谱分析时间对各组分保留时间窗口进行动态分配,仅当组分流出时才被采集。该分段方式不仅可以有效减少同时扫描不同离子对的数目,提高驻留时间、分析灵敏度和选择性,还可有效提升实验分析效率,适用于多种有机物的同时检测。闫君等[14]报道了动态多反应监测模式在食品领域的应用,但鲜有水质检测方面的报道。此外,LC-MS/MS在测定标准样品和待测样品时对不同基质带来的干扰较敏感,不同基质会对实验结果产生一定的误差,据文献[15-16]报道,选择合适的内标可减少不同基质带来的影响。
本研究拟建立直接进样-超高效液相色谱-串联质谱-动态多反应监测分析方法快速测定水中14种有机污染物。同时,使用上述14种同位素内标,通过对比分析标准样品与目标样品的保留时间和碎片离子,对目标样品进行分离及鉴定,使用内标校正法对各分析物进行定量测定。
1 实验部分
1.1 主要仪器与装置
Agilent 1290 Infinity Ⅱ超高效液相色谱仪(配备四元泵、自动进样器、柱温箱、二极管阵列检测器)、Agilent 6460三重四极杆质谱仪(配备Mass Hunter Acquisition Software B.01.08软件):美国Agilent公司产品;Milli-Q型超纯水机:美国Millipore公司产品。
1.2 主要试剂及材料
标准溶液:苦味酸有证标准溶液、13种混合有证标准溶液(丙烯酰胺、灭草松、2,4-滴、乐果、联苯胺、五氯酚、呋喃丹、甲萘威、阿特拉津、内吸磷、马拉硫磷、对硫磷、毒死蜱),浓度为100 mg/L,天津阿尔塔科技有限公司产品。
内标溶液:14种混合内标溶液(丙烯酰胺-D3、灭草松-D7、2,4-滴-13C6、乐果-D6、2,4-二硝基苯酚-D3、联苯胺-D8、五氯酚-13C6、呋喃丹-D3、甲萘威-D7、阿特拉津-D5、内吸磷-S-甲基、马拉硫磷-D6、对硫磷-D10、毒死蜱-D10),浓度为100 mg/L,天津阿尔塔科技有限公司产品。
主要试剂:乙腈(色谱纯),迪马科技公司产品;乙酸铵(色谱纯),德国Merck公司产品;超纯水(电阻率18.2 MΩ·cm),由Milli-Q超纯水系统制备。
1.3 标准溶液配制
标准储备液:分别移取100 μL 13种混合标准溶液和100 μL苦味酸单标溶液于10 mL棕色容量瓶中,用乙腈定容,配制成1 mg/L的14种混合标准储备液,于-18 ℃保存。
内标储备液:移取100 μL 14种混合内标溶液于10 mL棕色容量瓶中,用乙腈定容,配制成1 mg/L的14种混合内标储备液,于-18 ℃保存。
标准系列溶液:分别移取5、10、20、50、100、200、500、600、700、1 000 μL标准储备液和200 μL内标储备液于10 mL棕色容量瓶中,用水定容,得到0.5、1、2、5、10、20、50、60、70、100 μg/L 14种有机污染物的标准系列溶液,其中内标浓度为20 μg/L,现用现配。
1.4 水样采集与制备
按照HJ 164—2020[17]和HJ 493—2009[18]的相关规定采集和保存样品。用棕色玻璃瓶采集,当水样充满采样瓶加盖密封,4 ℃避光保存。水样经0.22 μm尼龙66滤膜过滤后,取1 mL置于棕色样品瓶,加入20 μL内标储备液,混匀后进样分析。
1.5 实验条件
1.5.1色谱条件 Agilent Poroshell 120 EC-C18色谱柱(3.0 mm×100 mm×2.7 μm);柱温40 ℃;流速0.3 mL/min;进样量10 μL;流动相:A为10 mmol/L乙酸铵水溶液,B为乙腈;梯度洗脱程序:0~0.5 min(95%A),0.5~3 min(95%~75%A),3~15 min(75%~35%A),15~20 min(35%~1%A),20~22 min( 1%A),22.01~25 min(95%A)。
1.5.2质谱条件 电喷雾电离源(ESI±);动态多反应监测模式;干燥气温度325 ℃,干燥气流速11 L/min;雾化器压力276 kPa;毛细管电压3 500 V;质谱参数列于表1。14种有机污染物的MRM色谱图示于图1。
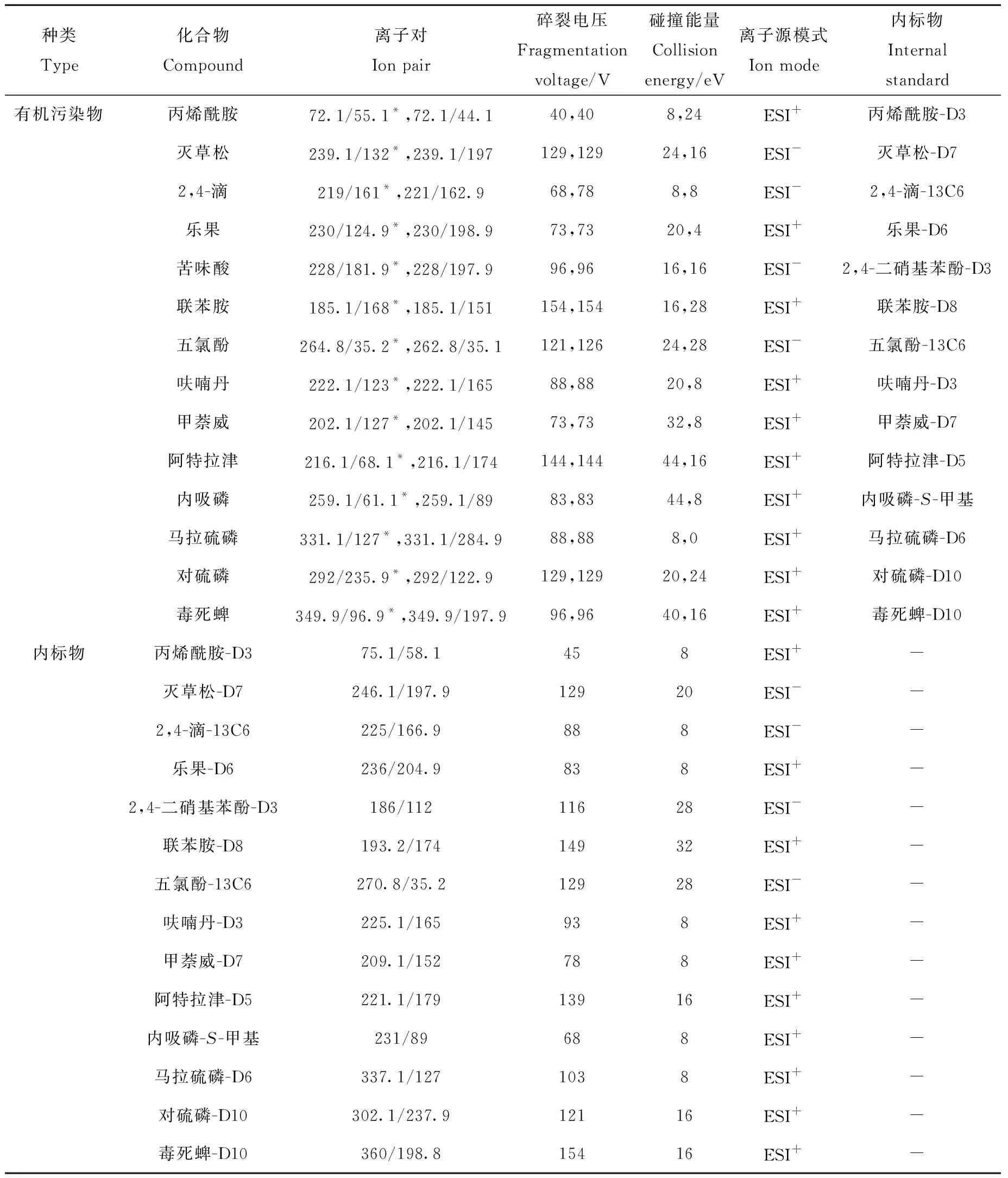
表1 14种有机污染物及其内标物的质谱条件Table 1 Mass spectrometry conditions of 14 organic pollutants and isotope internal standards
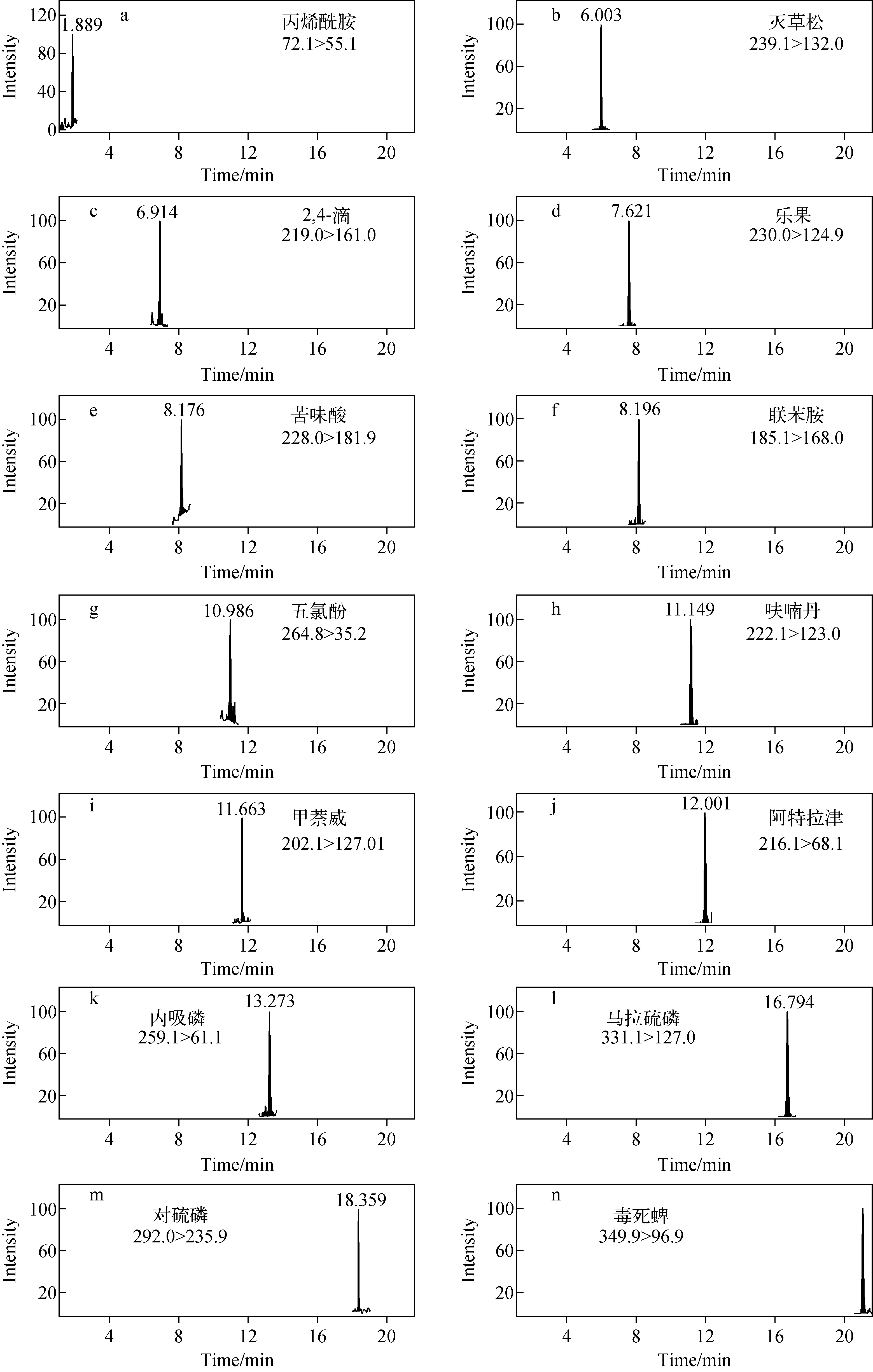
图1 14种有机污染物混合标准溶液的MRM色谱图(0.5 μg/L)Fig.1 MRM chromatograms of mixed standard solution of 14 organic pollutants (0.5 μg/L)
1.6 数据处理
采用Mass Hunter Qualitative Analysis和Quantitative Analysis软件进行数据的定性、定量处理,以样品的保留时间和定性/定量离子的相对丰度定性,根据定量离子的峰面积采用内标法定量。
2 结果与讨论
2.1 实验条件优化
2.1.1稀释液优化 通常,液相色谱在分离过程中存在“溶剂效应”,如果稀释液选择不合适,将影响色谱保留,导致色谱峰形异常,同时还会影响待测物的电喷雾电离效果,进而影响检测灵敏度。基于此,本研究分别以100%乙腈、50%乙腈-水溶液和纯水为溶剂,将混合标准溶液配制成20 μg/L稀释液进样分析,观察色谱峰形和分离效果。实验发现,随着稀释液中乙腈含量的增加,尤其是在纯乙腈体系中,灭草松、乐果、联苯胺、苦味酸等目标物及其内标物的色谱峰出现前沿峰;而使用纯水作为稀释液时,所得的色谱峰峰形对称且尖锐;其原因在于乙腈体系中样品与初始流动相的极性相差较大,存在强烈的“溶剂效应”,导致对目标物的洗脱效果和峰形都较差。因此,为消除由于“溶剂效应”引入的系统误差,同时与待测实际水样中的溶剂条件保持一致,本实验选择纯水作为混合标准溶液的稀释液。
2.1.2色谱条件优化 色谱柱的选择对于样品的分离非常重要,为确保各化合物在色谱柱上有较好的保留和分离效果,比较了Poroshell 120 EC-C18柱(3.0 mm×100 mm×2.7 μm)、Eclipse Plus C18柱(2.1 mm×100 mm×3.5 μm)和Eclipse Plus C18柱(4.6 mm×150 mm×5 μm)对14种有机污染物的分离效果。结果表明,使用Eclipse Plus C18柱(4.6 mm×150 mm×5 μm)时,流动相流量较大,与电喷雾离子化方式不匹配,导致离子化效率降低,质谱响应强度较低。相对于Eclipse Plus C18色谱柱(2.1 mm×100 mm×3.5 μm),Poroshell 120 EC-C18柱对各化合物具有较好的分离效果及峰形,原因在于该色谱柱装填了一种具有实心核和多孔外壳的表面多孔型填料,由于分析物在多孔外壳内外扩散速度更快,并具有非常均匀的填充柱床,所以这种色谱柱能够在2.7 μm填料的柱压下提供与亚2 μm填料相当的柱效,并且能够降低操作压力、改善峰形,适用于优化UPLC性能。因此,选用Poroshell 120 EC-C18色谱柱作为本实验的分析柱。
由于电喷雾质谱需在溶液状态下电离目标化合物,因此,流动相的选择要兼顾各目标化合物的色谱分离效果及质谱离子化效率。甲醇作为流动相存在共流出现象,信号之间相互干扰;相较而言,乙腈体系能够控制超高效液相色谱的系统压力,并获得较好的分离效果。本文分别考察乙腈-0.1%甲酸和乙腈-5 mmol/L乙酸铵作为流动相时14种有机物的分离效果。结果表明:选用0.1%甲酸能增加阿特拉津和甲萘威等化合物的响应值,但同时会导致联苯胺等化合物的色谱峰变形;选用5 mmol/L乙酸铵能明显改善各物质的峰形,大部分目标物的离子化效率高且峰形对称。综合考虑,最终选用乙腈-5 mmol/L乙酸铵作为流动相。
在梯度洗脱条件中,起始流动相通常采用高比例水相,有助于目标物与环境基体物质的分离,防止基质效应。本文以5 mmol/L乙酸铵水溶液(A)和乙腈(B)为流动相,比较方案1(0~0.5 min(95%A),0.5~20 min(95%~1%A),20~22 min(1%A),22.01~25 min(95%A))和方案2(0~0.5 min(95%A),0.5~3 min(95%~75%A),3~15 min(75%~35%A),15~20 min(35%~1%A),20~22 min(1%A),22.01~25 min(95%A))两种梯度条件下的分离效果。结果表明,方案1的色谱峰出峰时间集中在3~20 min,由于该区间化合物的结构和极性类似,各化合物的化学性质和色谱行为可能导致一些目标化合物出现共流出峰,进而影响峰形及其分离效果,使信号响应降低;方案2通过增加梯度,并放缓3~20 min梯度,各化合物依次出峰,分离效果好,色谱峰峰形良好,有助于提高检测灵敏度和准确度,故选方案2作为最佳梯度洗脱条件。
2.1.3质谱条件优化 根据各化合物的分子结构特征和化学电离性质,选择ESI源正、负离子模式分析。化学结构中含羧基类的酸性化合物容易失去质子带负电荷,一般采用负离子模式;化学结构中含氮的碱性化合物容易加和质子带正电荷,一般采用正离子模式。本研究中,苦味酸、灭草松、2,4-滴、五氯酚采用负离子模式检测,丙烯酰胺等10种化合物采用正离子模式检测。
不分段采集方式中多个MRM重叠会使灵敏度降低,而分段采集方式易降低分析方法通量。基于此,本研究采用动态多反应监测模式,按目标物保留时间设置监测离子扫描时间窗口,在不同时间段分别进行MRM扫描,以增加窗口期目标离子的扫描频率。该模式可提高质谱信号响应强度,增强质谱测定的稳定性和重现性,适合多种化合物的同时检测。
综上,本文采用正、负离子模式,动态多反应监测模式,优化后的离子对、锥孔电压、碰撞能量等参数列于表1。对14种有机污染物的混合标准溶液(0.5 μg/L)按照1.5节条件进样分析,14种有机污染物均有良好的响应。
2.2 方法学考察
2.2.1标准曲线的绘制 目前,水中有机物的定量分析多采用外标法[19-21],但存在测定仪器、样品基质带来的误差。本研究通过在水样中加入14种同位素内标,经内标校正,可有效避免误差,提高分析准确度,此外,还可通过内标物的峰形、峰面积监测样品与仪器的运行状态是否正常,同位素内标与目标物之间的对应关系列于表1。
按优化后的检测条件将14种化合物系列标准溶液按浓度由低到高的顺序依次进样测定,以待测物浓度为横坐标,待测物峰面积与相应同位素内标物峰面积之比(待测物峰面积/内标物峰面积)为纵坐标,得到的标准曲线示于图2,各化合物的线性范围、回归方程和相关系数列于表2。结果表明,各化合物在较宽的浓度范围内线性关系良好,相关系数R2均在0.999以上。
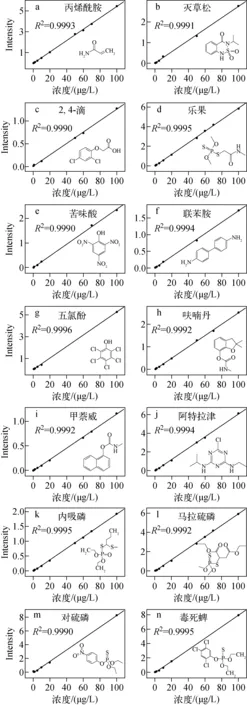
图2 水中14种有机污染物的标准曲线Fig.2 Standard curves of 14 organic pollutants in water
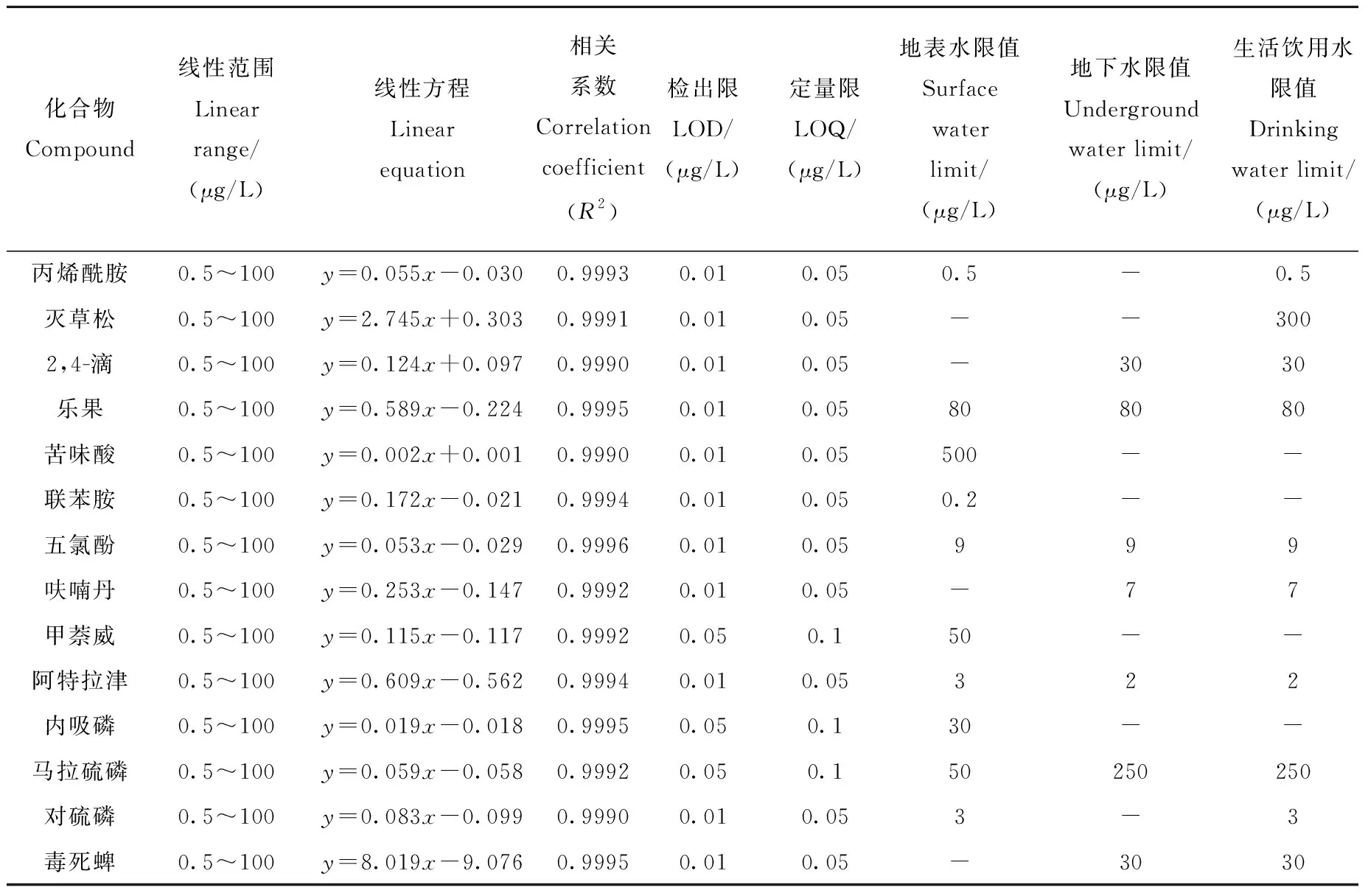
表2 水中14种有机污染物的线性范围、回归方程、相关系数、检出限、定量限和标准限值Table 2 Linear ranges, regression equations, correlation coefficients (R2), LODs, LOQs and standard limits of the 14 organic pollutants in water
2.2.2检出限与定量限 配制低浓度水平的14种待测物,以分子离子对的信噪比S/N≥3对应的质量浓度作为方法的检出限(LOD),以S/N≥10对应的质量浓度作为方法的定量限(LOQ),列于表2。14种待测物的LOD为0.01~0.05 μg/L,LOQ为0.05~0.10 μg/L,明显低于国家规定的浓度限值[3-5],能够满足直接进样测定地表水、地下水和生活饮用水中14种有机污染物的检测要求。
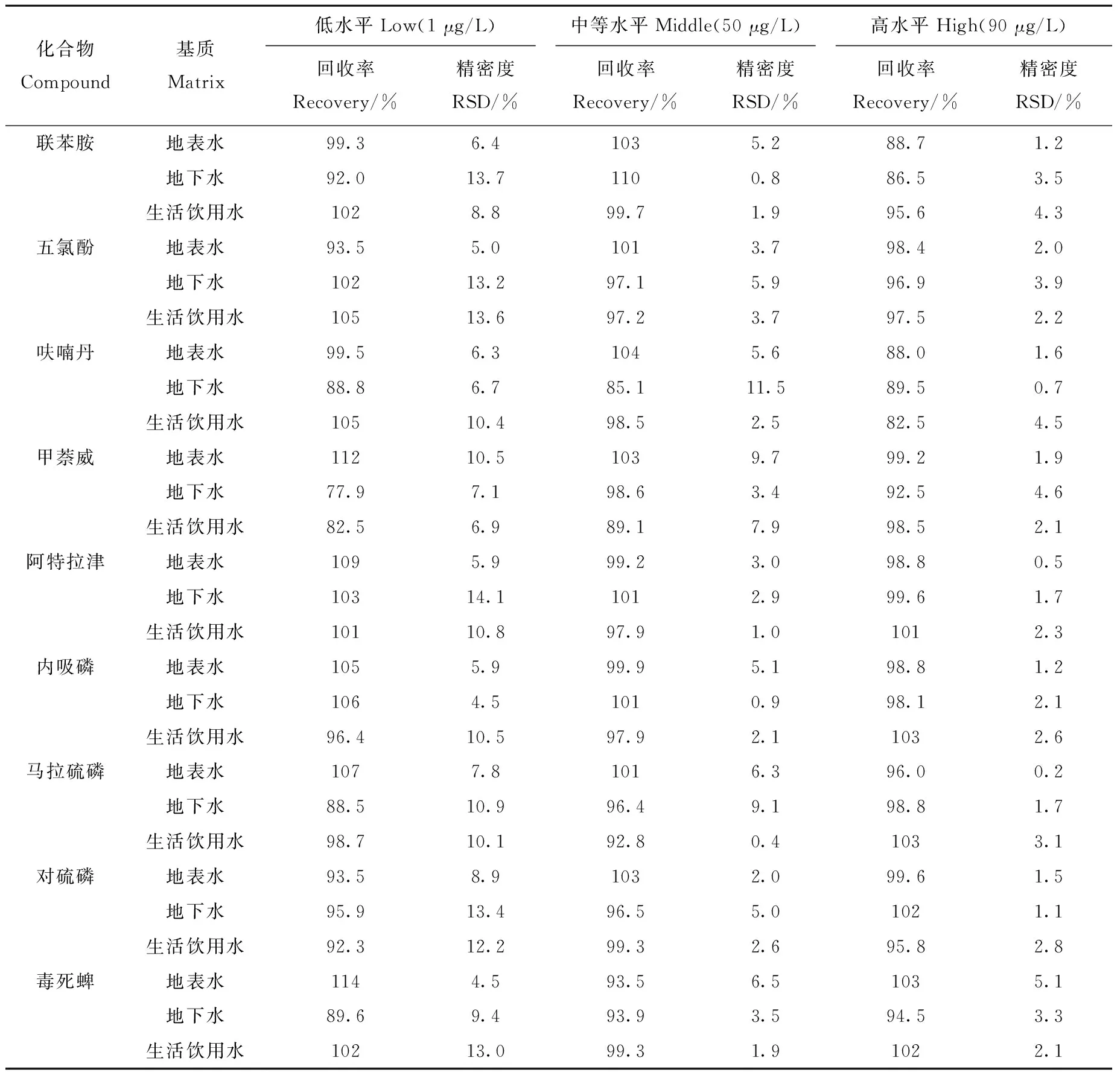
2.2.3加标回收率与精密度 选用不含14种待测组分的地表水、地下水和生活饮用水样品,分别添加低、中、高3个浓度水平的混合标准溶液,配制成浓度为1、50、90 μg/L的水样,每个浓度水平平行测定6次,分别加入20 μL内标储备液,测定加标回收率及精密度,结果列于表3。可见,在不同水体中,3个加标水平下14种有机污染物的平均回收率为77.9%~114%,相对标准偏差(RSD)为0.2%~14.1%,方法的准确性和精密度较好,能够满足环境监测质量控制要求。

表3 3个加标水平下,14种有机污染物的加标回收率和精密度(n=6)Table 3 Spiked recoveries and RSDs of the 14 organic pollutants at three spiked levels (n=6)
续表3
2.3 实际样品测定
采集北京城区地表水、地下水、生活饮用水3份水样,经滤膜过滤后加入内标,采用本方法对实际样品进行分析检测,结果在地表水样中检出了阿特拉津,其浓度为0.689 μg/L,远低于国家标准规定的浓度限值(3 μg/L)[3],示于图3,其余有机污染物均未检出。
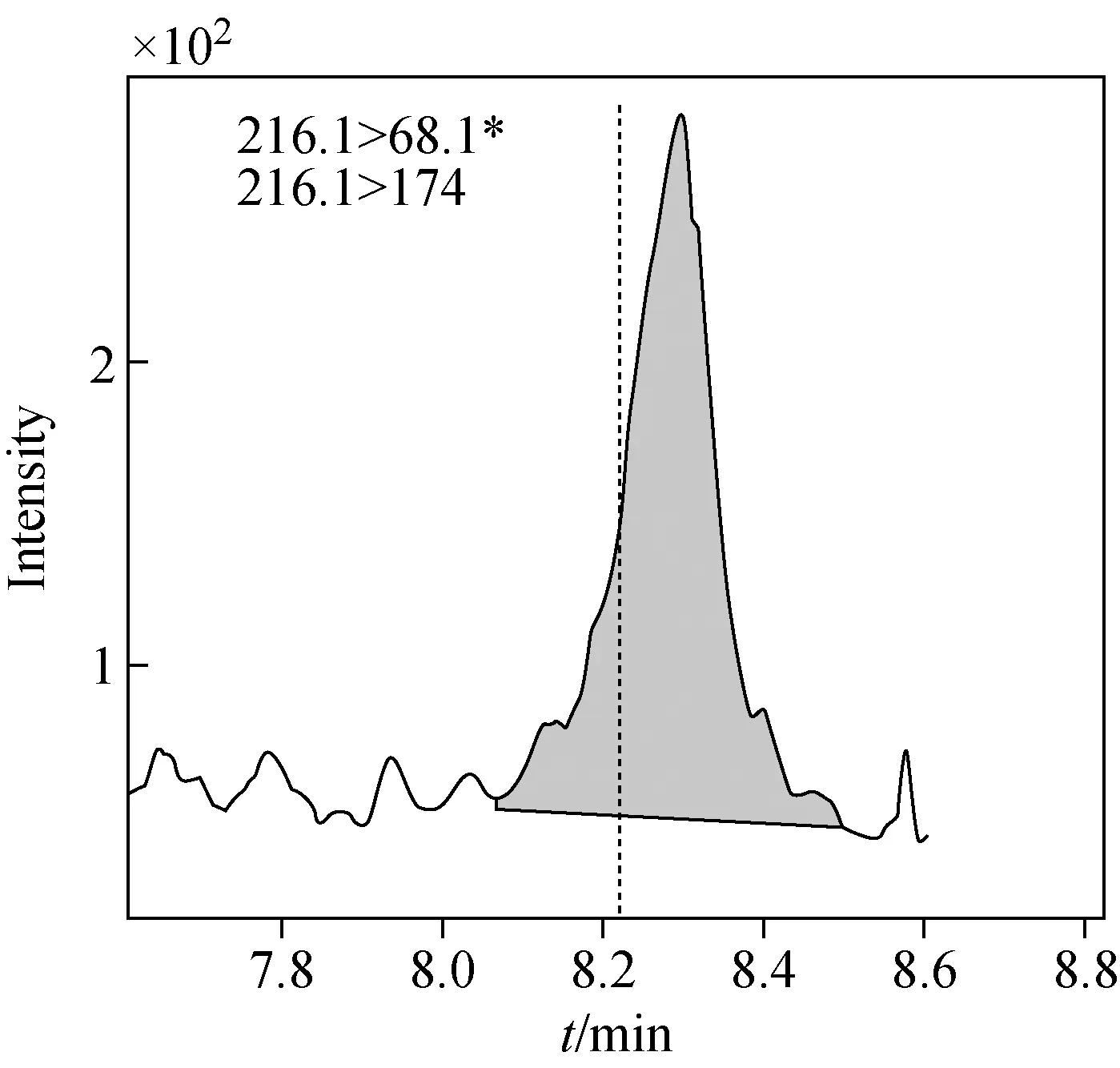
图3 阳性地表水样品中阿特拉津的MRM色谱图Fig.3 MRM chromatogram of atrazine in a positive surface water sample
3 结论
本文建立了一种基于电喷雾电离源正、负离子扫描,动态多反应监测、同位素内标定量分析的超高效液相色谱-串联质谱分析方法,用于测定水中14种有机污染物。该方法无需对水样富集,操作简便快速、灵敏度高,方法精密度和加标回收率能够满足国家标准的质量控制要求和限量检测要求,可为大批量同时快速检测地表水、地下水和生活饮用水中14种有机污染物提供技术参考。
