基于反应分子动力学的1-丁基-3-甲基咪唑硝酸盐与煤反应机理研究*
2022-03-14王兰云徐永亮
王兰云 刘 真 徐永亮 李 瑶 王 燕
(1.河南理工大学安全科学与工程学院,454003 河南焦作;2.煤炭安全生产与清洁高效利用省部共建协同创新中心,454003 河南焦作;3.河南省瓦斯地质与瓦斯治理重点实验室——省部共建国家重点实验室培育基地,454000 河南焦作)
0 引 言
采空区遗煤自燃是矿区治理的难点[1]。每年因煤自燃现象而产生的CO2量占总碳排放量的2%~3%[2-3],是实现碳达峰、碳中和目标亟待解决的问题。近年来,不少研究人员尝试将具有低熔点、低挥发性、环境友好、可回收等优点的离子液体(ionic li-quids,ILs)应用到防治煤自燃的研究中。WANG et al[4]研究发现ILs能够溶胀煤结构、溶解部分煤表面活性基团,从而抑制煤低温氧化。YANG et al[5]将长焰煤利用三种ILs作浸泡处理后进行了煤样氧化自燃放热和结构变化测试,认为对长焰煤自燃的抑制能力由强到弱的ILs依次为[Bmim],[Emim]和[Bmim][NO3]。肖旸等[6-7]研究发现咪唑类ILs处理能够增加煤分子成为活化分子的活化能,降低煤自燃反应速度。DENG et al[8]研究发现ILs在低温(温度低于130 ℃)时就可以破坏煤分子结构。
一般研究ILs影响煤自燃的实验步骤包括:混合、漂洗、过滤、干燥,但实际应用时ILs与煤混合后无法再分离,因此,ILs将与煤体共存,并在煤体氧化自燃过程中相互影响。为探究ILs与煤体共存时的燃烧特性,笔者曾使用锥形量热仪对ILs与煤混合后的样品进行燃烧性能测试分析,实验结果显示加入阴离子为-的三种ILs可以有效抑制煤的燃烧,但煤与不含氟元素的ILs(如[Bmim][Ac]和[Bmim][NO3])混合后,煤的燃烧速率比原煤的燃烧速率更快,达到放热量峰值的时间比原煤达到放热量峰值的时间更短。这一结果与之前报道的经ILs溶解溶胀清洗处理后,煤的自燃被抑制显然存在矛盾[5]。这就意味着利用溶解溶胀的处理方法与利用直接和煤混合的实验方法所得结果存在不一致之处,ILs存在于煤体会对燃烧测试分析结果产生重要影响,进而不利于准确评估ILs抑制煤燃烧的效果。另外,大部分相关报道还集中在经ILs溶解溶胀并清洗后的煤样的结构改变和自燃特性变化上,还未见针对混合样品的自燃尤其是在高温下燃烧机理的研究。可能存在低温阻燃效果较好的离子液体,但在环境温度到达某一温度时自发燃烧,反而促进煤体继续燃烧或分解生成更多有毒有害气体。因此,针对ILs是否能够用于抑制煤自燃,不仅应考虑低温氧化阶段的抑燃效果,还应考虑ILs长时间存在于高温煤体时两者的燃烧特性差异和相互反应过程。
由于ILs种类繁多且价格昂贵,通过传统实验方法来优选ILs存在成本高、过程复杂,不能全面获得燃烧可能产物种类等问题。探寻可行的反应动力学模拟(ReaxFF-MD)方法,揭示ILs促进或抑制煤自燃的机理,建立ILs与煤相互作用的反应分子动力学方法体系,可为优选ILs提供理论参考。CASTRO-MARCANO et al[9]使用ReaxFF-MD方法分析了煤的大分子模型燃烧过程中不同温度条件下产物的分布和反应物结构的变化,认为煤的燃烧过程是高温条件下煤热降解产生的小片段与O2,O2-及—OH等自由基反应的结果。BHOI et al[10]利用ReaxFF-MD方法对褐煤自燃及热解初始阶段进行研究,结果表明在褐煤的燃烧起始阶段,煤分子在高温条件下发生热解生成较小的碎片,而后裂解产物与氧气反应从而产生燃烧现象;而热解过程的起始机理则是氢自由基的脱氢反应。上述研究均说明ReaxFF-MD方法可对燃烧过程中反应物的结构变化和复杂中间反应进行合理表征。
[Bmim][NO3](1-丁基-3-甲基咪唑硝酸盐)能够溶解煤表面活性结构,且有研究[5]认为其具有抑制煤自燃的作用。笔者则从长远考虑,认为若注入的[Bmim][NO3]长期存在于煤体,有可能因ILs自身不稳定性、反应放热等引发煤分解燃烧,且燃烧后产生有毒烟气,发生与矿用高分子材料类似的事故[11]。此外,当外界环境发生变化如矿井发生火灾或爆炸事故时,用于抑制煤自燃的ILs就会成为新的危险源,特别是在采空区煤体的中心区域的ILs,由于与煤的混合且处于高温密封的环境中,ILs和煤分解产生的大量热量无法及时散出,有引发煤体持续分解并释放有毒有害气体的危险。本研究拟采用实验与模拟相结合的方法,首先采用FTT锥形量热仪对原煤及原煤-[Bmim][NO3]混合体系的燃烧性能进行测试,从宏观角度评价[Bmim][NO3]对煤燃烧过程的潜在影响;然后使用ReaxFF-MD方法从分子层面模拟分析高温无氧条件下[Bmim][NO3]阴、阳离子与煤之间的相互作用,揭示ILs对煤燃烧的影响机理,为ILs与煤共存的高温封闭区域的燃烧危险性分析提供有效的理论参考。为避免O2分子存在影响到分析ILs与煤相互作用的准确性,本研究暂不对O2存在条件下的反应过程进行模拟分析。
1 热重分析与FTT锥形量热仪实验
1.1 热重分析
实验煤样为河南义马烟煤,该煤发热量大,易发生自燃。将煤块粉碎、研磨,并选取粒径为75 μm~180 μm的新鲜煤样,于室温真空干燥24 h,密封保存。[Bmim][NO3](C8H15N3O3)从中国科学院兰州化学物理研究所购得,密度为1.16 g/cm3,纯度为99%。采用德国NETZSCH STA449C热分析仪,实验用煤的质量及[Bmim][NO3]的质量均为5 mg,温度范围为30 ℃~800 ℃,升温速率为10 K/min,实验过程中持续通入100 mL/min的高纯氮气(φN2>99.99%)。
1.2 FTT锥形量热仪实验
FTT(fire testing technology)锥形量热仪实验按照ISO 5660(GB/T 16172-2007)标准进行,锥形量热仪热辐射释放通量设定为50 kW/m2。实验用煤样和[Bmim][NO3]均与热分析实验一致;实验开始前将煤样与[Bmim][NO3]按质量比为1∶1混合后密封放置1 h,以保证煤与离子液体充分混合。完成仪器常数C-Factor校正后准备进行实验,实验前在10 cm×10 cm×10 cm的样品盒内铺设一层锡纸以使样品受热均匀,样品台高度设置为10 cm,称量20 g样品均匀平铺于样品盒内。待仪器热辐射通量达到设定值后开始标定基线,然后打开隔热板进行点火实验,并记录实验数据。
1.3 实验数据分析
图1a所示为通过热分析实验分别得到的烟煤和[Bmim][NO3]的热重曲线。由图1a可知,与烟煤相比,[Bmim][NO3]的热分解温度明显较低,为296.6 ℃,而烟煤需达到334 ℃时才开始发生热分解,说明[Bmim][NO3]的热稳定性明显弱于烟煤的热稳定性。图1b所示为通过FTT锥形量热仪实验分别得到的烟煤及烟煤-[Bmim][NO3]混合物的单位面积热释放速率曲线。由图1b可以看出,与烟煤相比,加入[Bmim][NO3]的混合体系在燃烧时单位面积热释放速率达到峰值的时间明显前移,且存在两个峰值。推测P1为[Bmim][NO3]首先燃烧并达到单位面积热释放速率的峰值,[Bmim][NO3]提前分解为煤燃烧提供初始热量,使得煤燃烧放热加速,从而出现P2峰值,由此可见,[Bmim][NO3]的加入对煤的燃烧进程具有加速作用。
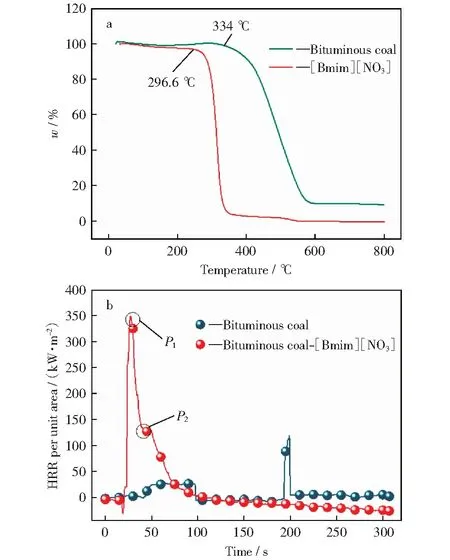
图1 纯烟煤和[Bmim][NO3]的TG曲线及纯烟煤和烟煤-[Bmim][NO3]混和物在FTT测试中单位面积热释放速率(HRR)曲线
2 煤-[Bmim][NO3]体系反应分子动力学(ReaxFF)模拟
2.1 ReaxFF反应力场
ReaxFF-MD利用键序的概念来描述系统中每个原子的相互作用,系统中的总能量(Esystem)包含以下几个部分:共价键键能(Ebond)、过饱和键补偿能(Eover)、不饱和补偿能(Eunder)、孤对电子能(Elp)、键角能(Eval)和扭转能(Etors),以及非键作用的库仑能量(ECoulomb)和范德华能量(EvanWaals);并利用键断裂和形成过程中键级的变化描述真实结构模型中的化学反应过程[12]。
Esystem=Ebond+Eover+Eunder+Elp+Eval+Etors+ECoulomb+EvanWaals
(1)
在ReaxFF分子动力学模拟中,力场文件的选择对模拟结果的准确性有较大的影响。现有的力场主要分为两个分支体系:燃烧分支和水分支。两者的不同在于H元素和O元素参数的设置上。在燃烧分支的力场文件中水主要以气相形式存在于反应体系,而在水分支的力场文件中以水为液态形式进行参数设置。
本研究模拟的反应环境为高温条件,因此选择燃烧分支的相关力场。BUDZIEN et al[13]在C/H力场[14]的基础上增加了O元素和N元素的参数设置,开发出包含了C,H,O,N四种元素的反应力场,并将其应用于季戊四醇四硝酸酯的燃爆性能模拟过程中,得到了较好的实验结果。因此,本研究使用该力场参数进行煤燃烧模拟计算。
2.2 计算模型的构建和计算
煤的结构较为复杂,包含大量的化学键和官能团[15]。近十年来,研究者们发展了一系列成熟的方法来构建煤的大分子结构。到2012年底,已经构建出超过134种煤的大分子模型[16]。根据NAKAMURA et al[17-19]提出的烟煤、褐煤和无烟煤的典型大分子结构,可知煤结构中的主要重复单元为带有含氧官能团支链的芳香结构。而LI et al[20]研究指出,煤的燃烧性能与羧基的含量具有较强的相关性,其中褐煤的羧基含量在几种煤样的羧基含量中是最高的。因此,本研究选用带有羧基官能团的多环结构(C14H10O2,如图2所示)作为煤分子的模型。利用Material Studio软件中的Amorphous模块构建煤分子与[Bmim][NO3]混合体系模型(无定型单元为21.341 nm×21.341 nm×21.341 nm的立方体结构),包含10个煤分子与10个[Bmim][NO3],初始密度为1.21 g/cm3。使用Material Studio软件中的Frocite模块进行结构弛豫,力场选用CommpassII,由于体系不大,将精度调节为Fine。先选择NVT系综(温度(T)、体积(V)、原子数(N)恒定)将随机分布的分子结构充分地分散开,然后在298 K~1 500 K条件下进行退火处理,系综保持不变。经过10次循环退火后,再将退火完成后的模型在NPT系综(温度(T)、压强(p)、原子数(N)恒定)条件下进行分子动力学运算。温度设置为1 200 K,压力为1 GPa,运算时间为200 ps。最终得到密度为1.45 g/cm3的初始模型,如图2所示。
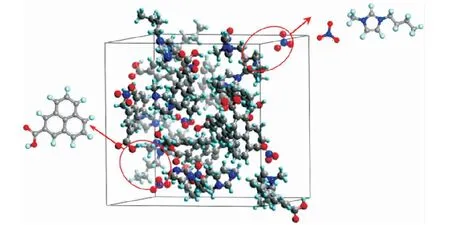
图2 退火完成后煤-[Bmim][NO3]混合体系的3D模型
为分析高温环境下[Bmim][NO3]与煤的相互作用,将使用Material Studio软件完成优化的初始模型文件(.pdb格式)导入Lammps[21]中使用并进行ReaxFF-MD运算,分别在1 200 K,1 600 K,1 800 K和2 200 K四个温度条件下进行模拟计算。系综类型为NVT系综,步长为0.25 fs,间隔1 ps记录一次数据,控温方式为Berendsen,每组运行时间为1 ns。
由于反应分子动力学方法运算时间较长,当模拟时间与实际实验时间在不同数量级上时,提高模拟温度可以有效增加原子之间的碰撞从而提升反应效率。因此,整个模拟过程温度较实际过程温度更高。但是,根据阿仑尼乌斯公式[22]及ZHENG et al[23]研究结果可知,升高温度会加快化学反应速率,对活化能的影响却不明显,因此其对反应机理并不产生影响,不会影响结果的准确性。
3 结果与讨论
3.1 反应路径统计与分析
图3所示为1 200 K,1 600 K,1 800 K和2 200 K四个温度条件下C14H10O2,[Bmim]+,[NO3]-的分子数目随模拟时间的变化规律。由图3可以看出,除1 200 K外,其余三个温度条件下煤分子数目在7 ps内都降到了0,当温度为1 200 K时,煤分子数目下降过程大致分为三个阶段:0 ps~10 ps内分子数目下降50%,10 ps~40 ps内分子数目下降20%,40 ps~70 ps内分子数目下降10%,70 ps~200 ps内分子数目下降10%,且随着反应的进行,消耗相同数目的煤分子需要更长的时间。YAN et al[24]利用Reax-FF研究了褐煤的氧化过程,研究发现当计算温度设置在低温区(1 000 K~1 500 K)时,体系中的褐煤分子没有发生氧化,发生的是脱羧反应,而在模拟后期由于羧基数目减少导致煤分子的氧化反应速度减缓。
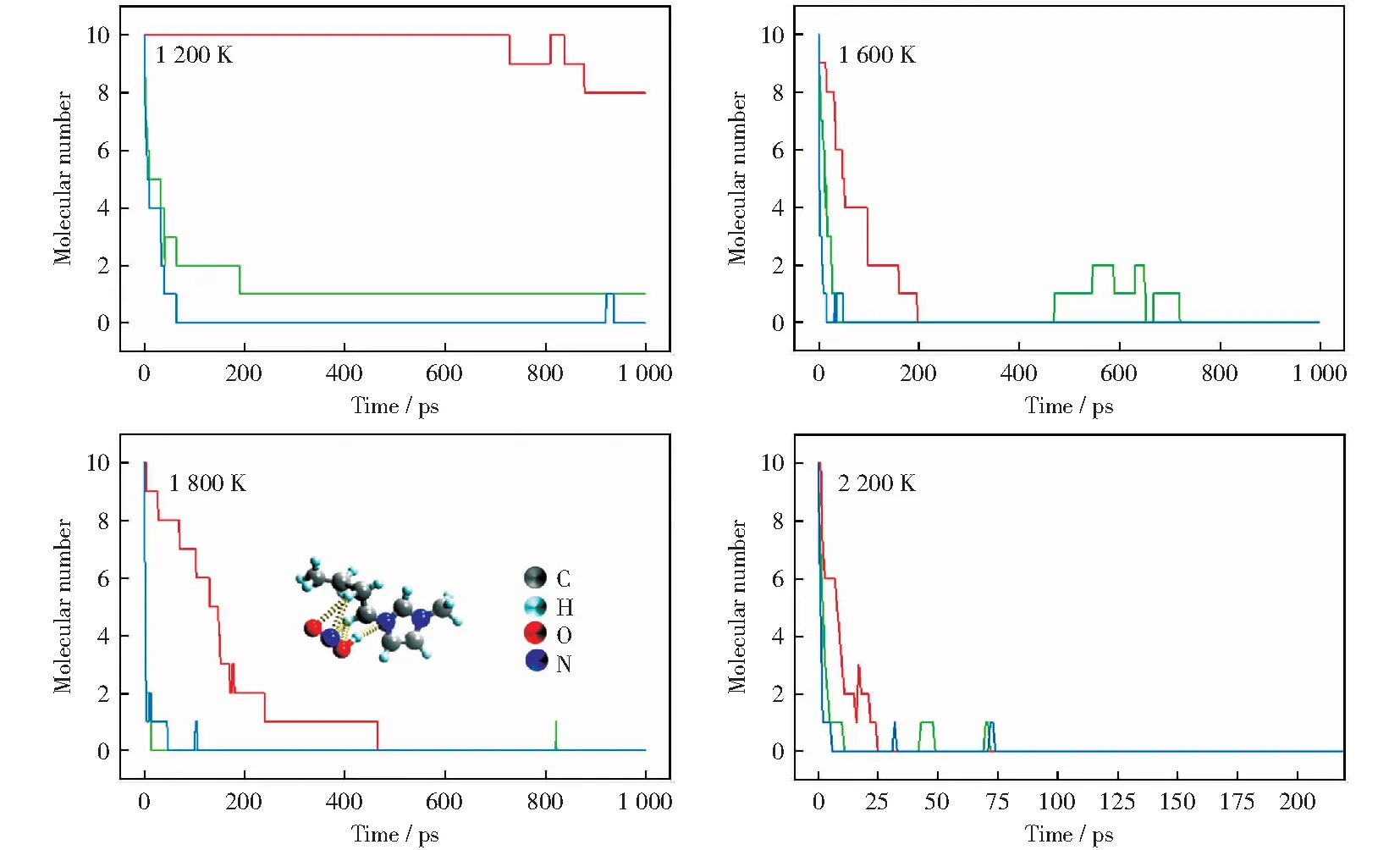
图3 Reax-FF模拟中不同温度条件下反应物分子数目随时间的分布
[Bmim][NO3]中的阴、阳离子在不同温度条件下的消耗规律差异较大。阴离子对温度的敏感程度较大,在各个温度阶段消耗速率都比较大,而阳离子在1 200 K时保持了相对稳定的状态,在0 ps~724 ps时间段内分子数目保持不变,仅有20%在计算后期参与反应。当温度升高(1 600 K~2 200 K),[Bmim]+消耗的速率逐渐加快,说明温度与[Bmim]+的消耗存在明显正相关性。当温度上升至2 200 K时,[Bmim]+在30 ps内便消耗殆尽。值得注意的是温度在1 800 K时[Bmim]+的消耗情况。由图3b和图3c可知,当温度为1 600 K时,体系内[Bmim]+约在200 ps内便已全部参与反应,但当温度升高至1 800 K时[Bmim]+参与反应的速度反而下降。分析温度为1 800 K时体系内的产物发现,在该温度条件下[Bmim]+与·NO2形成了氢键(见图3c)。赵亚梅等[25]曾对[s-Bmim][PF6]的热稳定性进行研究,发现增强分子间氢键有助于提高体系的热稳定性。这可能是[Bmim]+保持稳定的原因。
图4a所示为1 200 K~2 200 K的温度条件下,煤与[Bmim][NO3]中的[NO3]-的反应路径统计。由图4a可知,煤主要与[Bmim][NO3]中的[NO3]-发生反应生成HNO3,但产生的HNO3并不能稳定存在,而是继续反应生成氮氧化物。当温度升高至1 600 K时,煤与部分[NO3]-反应会直接产生NO2等氮氧化物。在1 800 K时,[NO3]-直接与煤的反应进一步减少,反应更多集中于煤直接与氮氧化物的反应。在2 200 K时,煤的反应途径变得更加复杂,大致可以分为三类:1) [NO3]-与煤的反应;2) 氮氧化物与煤的反应;3) 煤的自身分解反应。相较于低温阶段,当温度为2 200 K时,煤与[NO3]-的反应类型数增加,但与氮氧化物的反应类型数反而下降。此外,有大约50%的煤分子发生了自分解反应,这一反应途径在所有温度条件下占比均为最大。
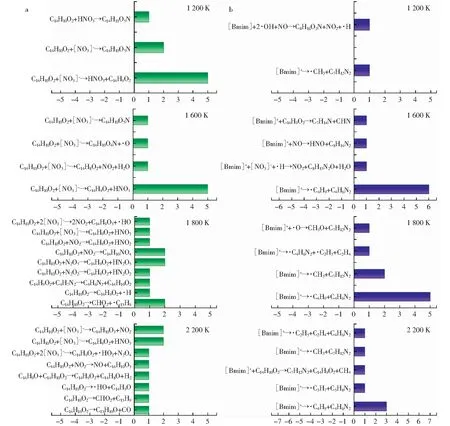
图4 不同温度条件下煤与[Bmim][NO3]反应路径统计
图4b所示为1 200 K~2 200 K的温度条件下煤与[Bmim]+的反应路径统计。当温度为1 200 K时,[Bmim]+的活跃度较低,仅有20%参与反应,且主要是支链断裂产生·CH3的过程(见图5)。OHTANI et al[26]利用气相色谱及质谱分析法研究了阴离子为卤素原子的ILs的热稳定性影响因素,结果表明,[Bmim]+支链上远离N原子的C首先脱落形成·CH3。当温度升高至1 600 K,咪唑环上的支链进一步裂解产生·C4H9。当温度在1 800 K~2 200 K时,低温阶段生成的烷基自由基中β位发生裂解反应产生不饱和的烯/炔烃类物质,如C2H4和·C4H9。CHEN et al[27]利用量子化学(QM)的方法对3-甲基-1-庚基在500 K~2 500 K条件下的热解过程进行了研究,计算发现高温会使长链自由基转化为不饱和烃类,低温反应过程产生的大量烷烃自由基为高温条件下不饱和烃类的产生奠定了重要基础,该结论与本研究的ReaxFF-MD计算分析结果一致。
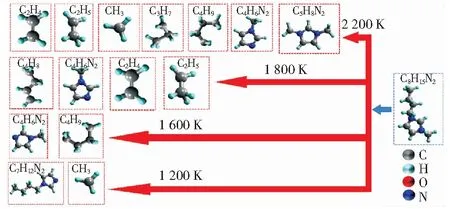
图5 ReaxFF模拟中不同温度条件下[Bmim]+反应后产物统计
低温条件下,离子液体中的[NO3]-与煤分子中的羧基发生反应,生成HNO3或氮氧化物,煤分子则变为易得电子结构,反应活性增强;[Bmim]+支链上离咪唑环较远的C原子脱落,形成小分子烷基。高温条件下,煤分子倾向于发生自分解反应,[NO3]-对其产生的影响减弱;而[Bmim]+热解程度加深,产生大量小分子有机气体,如C2H4,·C4H9等。
3.2 [Bmim][NO3]与煤的反应产物分析
3.2.1 CO/CO2
CHEN et al[28]运用反应分子动力学方法研究了贫氧条件下褐煤的自燃过程,发现在1 000 K条件下几乎没有CO和CO2产生。当温度升至1 500 K时,CO开始产生,说明CO的生成与温度有关。图6所示为本研究所得不同温度条件下CO和CO2及—COOH的分子数目随时间的变化曲线。当温度为1 200 K时,体系中没有CO和CO2产生,说明该温度不满足生成CO或CO2的条件。通过分析反应物消耗路径可以发现,[NO3]-虽然可以夺取羧基中的H原子,从而形成—COO,但该温度条件下反应系统无法提供C—O键断裂所需的能量,导致产生CO的过程中断。当温度升高至1 600 K,CO和CO2开始出现,但此时两者产生的时间较短,分子数目较少,且初始生成时间较长。当温度在1 800 K左右,CO和CO2的产生时间均显著提前,CO初始出现时间早于CO2初始出现时间,但分子数目少于CO2的分子数目。当温度达到2 200 K时,CO和CO2在10 ps内便开始生成,但之后的变化趋势却明显不同。高温状态下生成的CO2的分子数目少于CO的分子数目,CO2在反应后期逐渐消失,而CO分子数目则持续增加,说明高温更有利于热解体系产生CO。此外,由图6b可以看出,随着羧基分子数目的减少,CO2的分子数目开始增加,在1 600 K~1 800 K温度范围内,这一趋势较为明显,说明CO2的分子数目与羧基(—COOH)的分子数目具有较强的相关性。
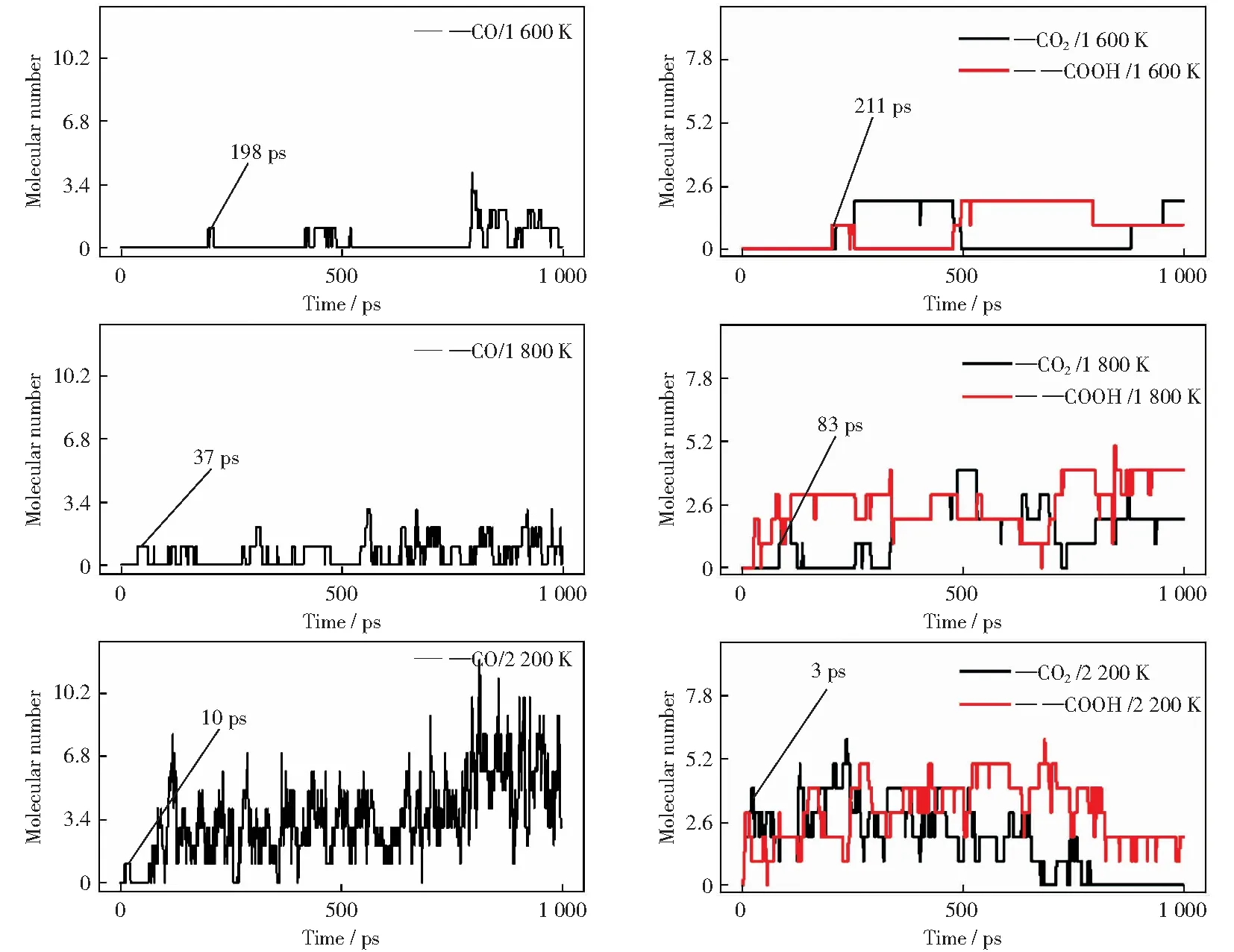
图6 ReaxFF模拟中不同温度条件下CO和CO2及—COOH的分子数目随时间的分布
图7所示为煤-[Bmim][NO3]混合体系热解形成CO机理。由图7可以看出,当温度在1 600 K时,CO主要是通过反应(a)和反应(b)两类反应产生的,其中反应(a)中的R—C—O—C—O—结构是反应初期由煤分子与[NO3]-反应生成的易得电子结构,反应(b)是[Bmim]+的二级反应过程,其反应物为[Bmim]+支链断裂产生。值得注意的是,反应(b)在反应发生10 ps后出现了可逆反应,这说明在该温度条件下反应(b)的稳定性比反应(a)的稳定性弱。
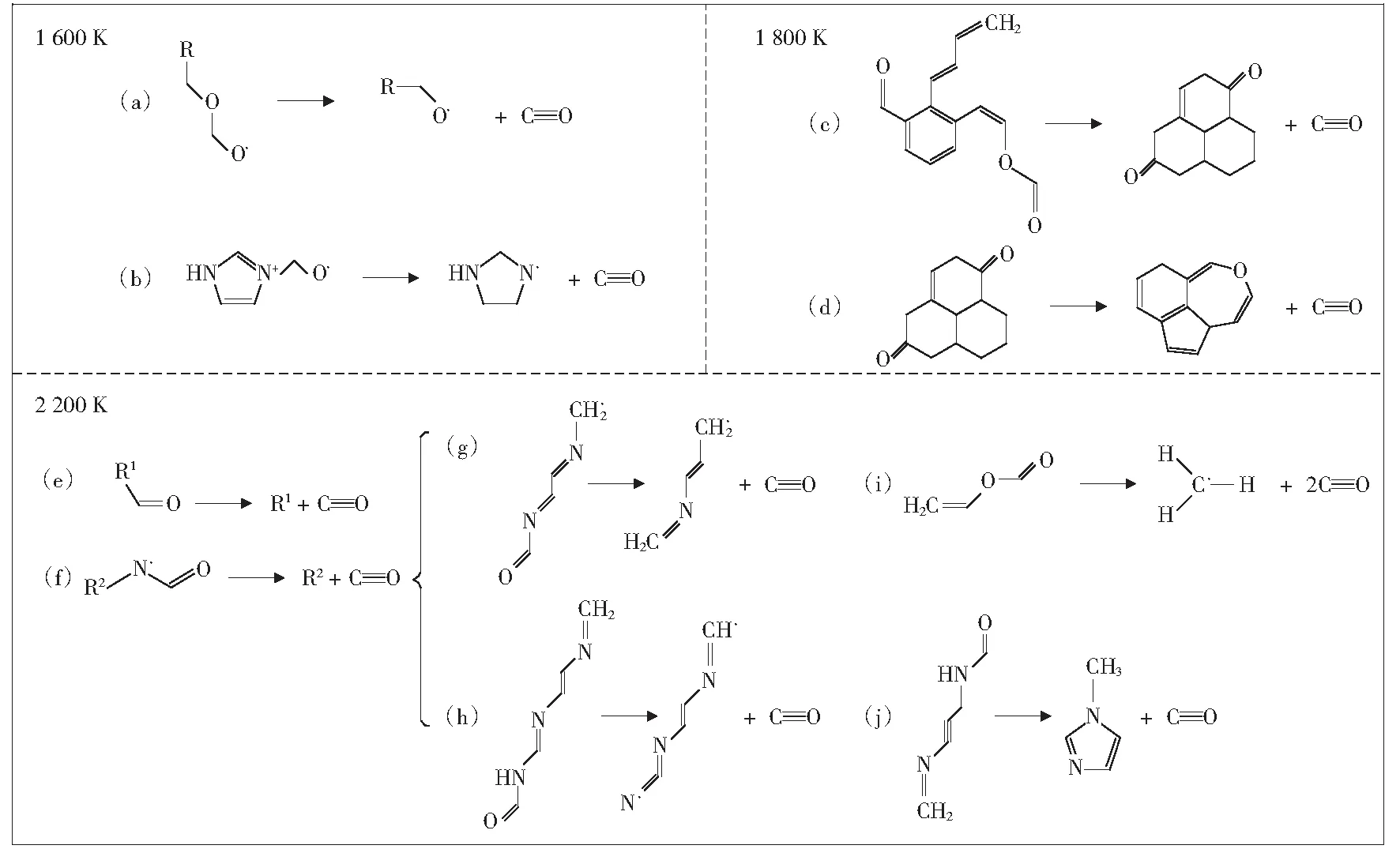
图7 煤-[Bmim][NO3]混合体系热解形成CO机理
当温度为1 800 K时,除在1 600 K条件下发生的反应(a)和反应(b)两类反应以外,还出现了支链重新闭合成环反应(c)。当反应物中最长的含氧支链断裂形成CO后,原本已经断裂的芳香环再次闭合,形成含有醛基的芳香环结构。新形成的产物通过反应(d)产生CO,在反应(d)中,反应物中一个氧原子从支链上脱离形成CO,另一个则从支链转移至环上与相邻碳原子形成醚键。
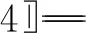
图8所示为煤-[Bmim][NO3]混合体系热解形成CO2机理。由图8可以看出,当温度在1 600 K~1 800 K时,CO2主要通过反应(a)产生。反应(a)为可逆反应,其正反应过程如反应(b)所示,羧基脱去H后R—C键断裂,产生CO2。逆反应过程如反应(c)所示,当游离的CO2遇到H离子或能够产生H离子的物质时,CO2会再次结合H重新形成羧基。这也说明羧基是生成CO2的主要反应物,符合其他学者[29]发现的“煤中羧基碳物质的量比越大,CO2产量越大”的规律。
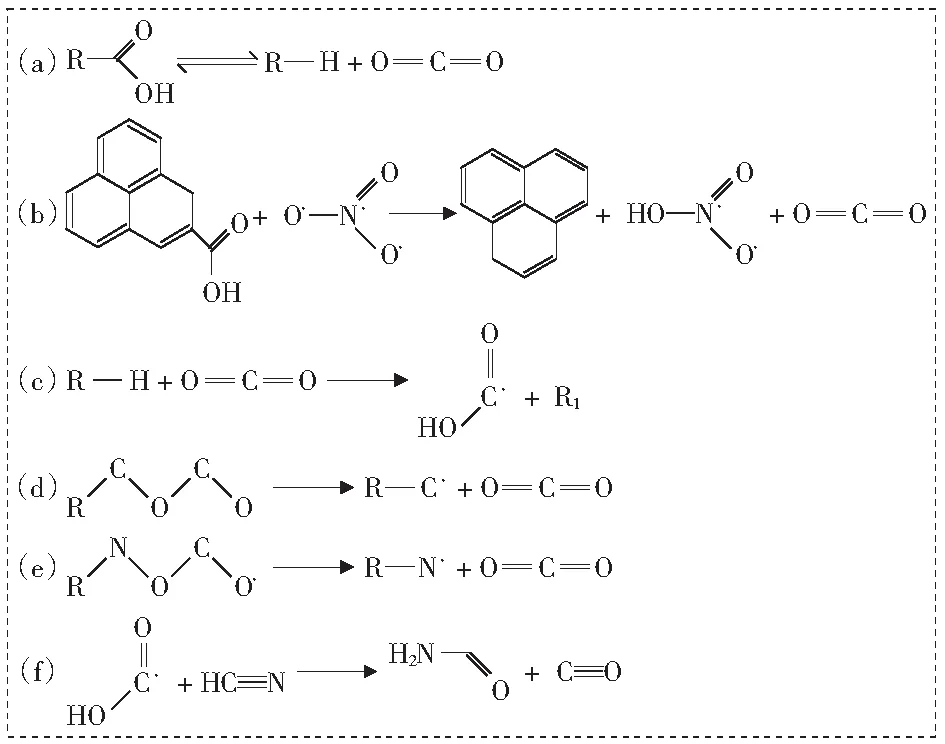
图8 煤-[Bmim][NO3]混合体系热解形成CO2机理
但当温度升高至2 200 K时,羧基的减少与CO2的生成不再保持一定的对应关系,因为该温度条件下CO2的产生不再依赖于脱羧反应。由反应(d)和反应(e)可知,—O—C—O—结构中C—O键的断裂也会产生CO2;另外,部分羧基开始反应产生CO,如反应(f)中羧基与HCN反应脱去—OH产生CO的过程。
显然,加入[Bmim][NO3]对CO与CO2的产生有着明显的影响。较低温度条件下[NO3]-与煤中的羧基反应,羧基失去·H后形成的—C—O—C—O结构会继续反应产生CO与CO2。当温度逐渐升高,[Bmim]+裂解程度逐渐变大,产生的小分子碎片如—N—C等与含氧自由基反应生成CO与CO2。
3.2.2 HCN/NH3
通过对产物进行统计可以发现,反应中生成的产物主要包括H2O、氮氧化物、小分子烷烃与烯烃、HCHO、NH3、HCN。这与BHOI et al[10,28]在相似条件下模拟煤热解过程所得结果一致,NELSON et al[30-31]运用实验方法也同样检测到CO,CO2,HCOH,NH3等产物的生成。
相较于纯煤,当煤样与[Bmim][NO3]混合后,生成物产生时间和产生过程发生了较大改变。图9所示为1 200 K~2 200 K四个温度条件下主要产物的分子数目随时间的变化趋势。由图9可以看出,当温度为1 200 K时,主要产物为H2O与NO2,其中NO2的分子数目并不如H2O的分子数目稳定,数目波动较大,这说明NO2作为中间产物频繁参与反应。当温度升高至1 600 K时,NO2逐渐转化为NO,且体系中开始出现小分子烃类,C2H4的含量随着反应时间逐渐升高。温度升高至1 800 K,体系内的主要产物开始出现分化,H2O,HCHO,C2H4的含量远高于其余几种产物的含量,这说明在该温度条件下这几类产物已经可以稳定存在。当温度为2 200 K时,H2O的分子数目继续上升,CH4的分子数目出现快速增长,这说明高温使反应物上的碳链快速裂解,小分子烷基与·H结合形成了CH4。此外,HCHO在1 800 K时能够稳定在一定的数目,但当温度升高至2 200 K时却出现较大幅度的减少,这说明HCHO在该温度条件下参与反应,同时生成了HCN与NH3等新的产物。
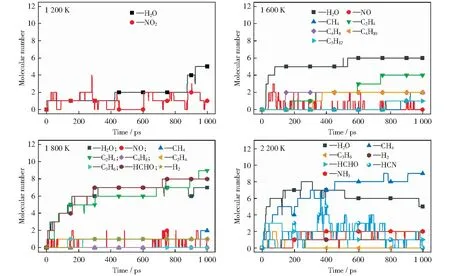
图9 ReaxFF模拟中不同温度条件下主要产物的分子数目随时间的分布
表1所示为不同温度条件下的生成物统计结果。反应生成物可以划分为四类,即无机物、碳原子数目小于4的小分子化合物(
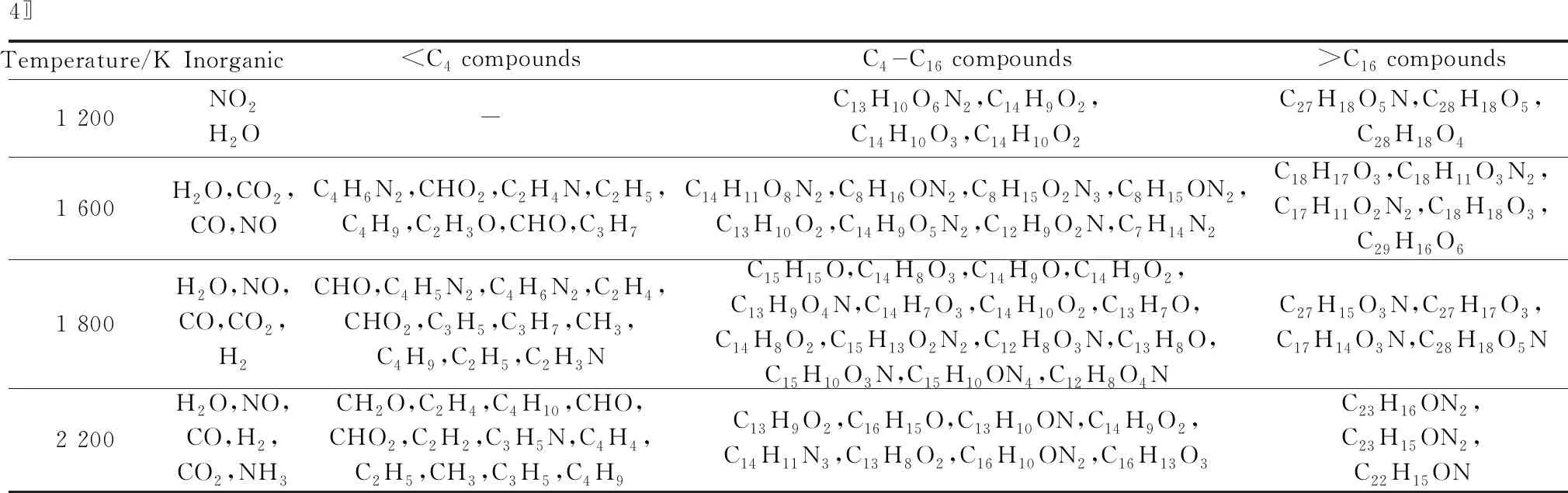
表1 不同温度条件下的生成物统计结果(Cn表示碳原子数目为n的物质)
此外,通过对碳原子数目为4~16的生成物(C4~C16)和碳原子数目大于16(>C16)的生成物的元素种类进行分析后发现,这些产物中多数含有N元素,而原煤分子模型构建过程中并未添加N元素,这说明[Bmim][NO3]的加入促进了中间产物的生成,提高了反应速率,而小分子化合物增多会导致放热量增大。
图10所示为生成HCN的主要反应路径,表2所示为图10中涉及的化学反应。煤分子与[Bmim]+通过反应R1和反应R2脱去支链上的碳氢原子。其中,反应R1为[Bmim]+与煤分子反应脱去甲基产生CH4与·C7H12N2的过程,反应R2为[Bmim]+支链断裂生成C4H6N2的过程。反应R1和反应R2中的产物经过反应R3和反应R8,咪唑环支链进一步发生断裂,生成·C3H3N2。大约在480 ps开始形成HCN。HCN主要是通过R5和R6两种反应途径分解产生,其中反应R6中的反应物由·C4H6N2中咪唑环断裂而来。反应R7和反应R8为可逆反应,由于甲基受咪唑环中电负性较强的N原子影响,容易在生成游离基团后再次与N原子键合。如图10所示,[Bmim]+中咪唑环断裂后产生HCN,而过高的温度是造成咪唑环分解的重要原因。
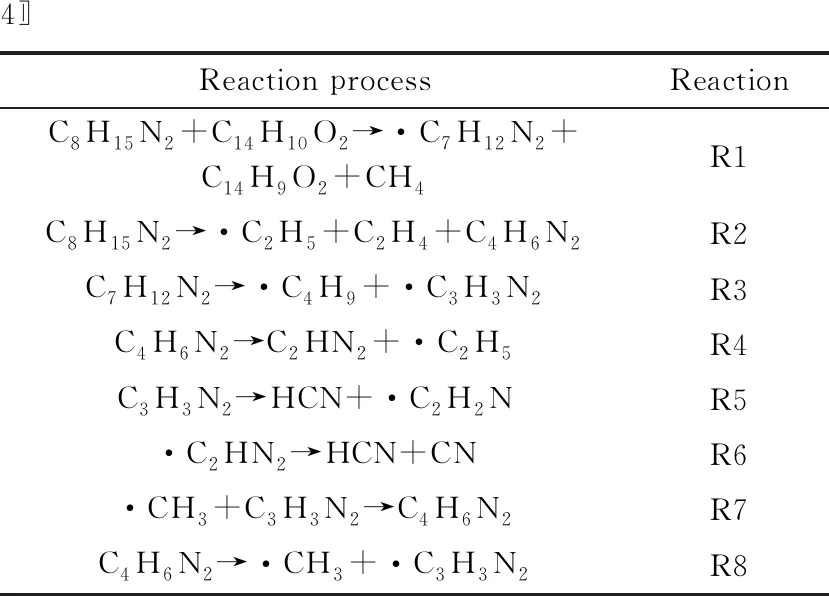
表2 图10中涉及反应过程
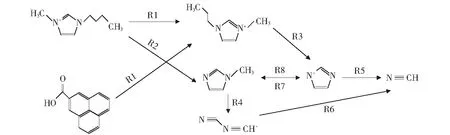
图10 生成HCN的主要反应路径(箭头指向生成物,R1~R8为反应序号)
NH3是体系中N的另一种转化形式。相较于HCN的形成过程,NH3的形成路径相对简单。如图11所示,氨基与·HNO及·HCO通过R1和R4途径产生NH3。结合图12a可知,氨基与·H结合形成NH3。而·N2H与体系中·H和·OH通过反应R2生成NH3,其原子转移过程如图12b所示。·N2H中与H相连的N通过结合游离状态的H与OH中的H结合形成NH3。而反应R3是NH2与羧基中的H结合产生NH3。R4的反应机理与R1的反应机理相同。图11中涉及的反应过程见表3。
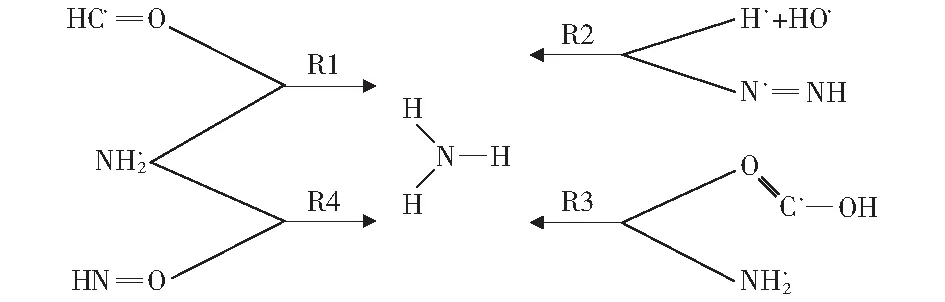
图11 生成NH3的主要反应路径
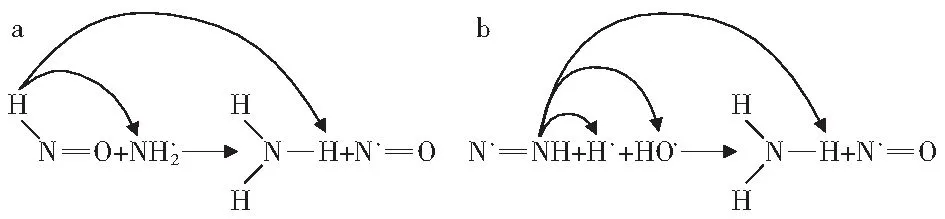
图12 NH3生成过程中原子迁移过程
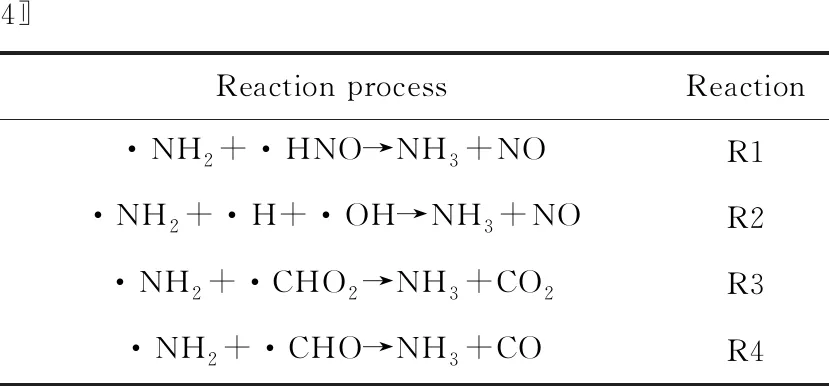
表3 图11中涉及的反应过程
4 结 论
1) FTT锥形量热仪实验结果表明,与纯煤相比,[Bmim][NO3]先于煤发生分解放热,煤中[Bmim][NO3]的存在能够增加体系热释放总量。
2) [Bmim][NO3]的添加会使煤的单位面积热释放速率(HRR)达到峰值的时间提前。
3) 低温条件下[Bmim][NO3]中阴离子与煤分子侧链上羧基反应形成得电子结构,从而增强煤的反应活性。随温度升高,阳离子中的烷基侧链和咪唑杂环依次分解,生成大量自由基,使煤的反应过程加快。
4) [Bmim]+支链断裂形成的自由基能够促进煤结构中脂肪结构和芳香结构分解产生CO;其杂环断裂后形成的N—C结构与氮氧化物反应产生的—N—C—O—C—O还会增加CO2的生成量。另外,当氧气浓度不足时,NH3和HCN无法转化为氮氧化物,对人和环境的安全造成极大的威胁。
