XPO4(X=Lu,Y)电子结构的第一性原理研究
2022-03-04文杜林熊明姚
文杜林,熊明姚,张 苗,苏 欣
( 伊犁师范大学 新疆凝聚态相变与微结构实验室,伊宁 835000)
1 引 言
磷酸盐晶体具有结构稳定,非线性光学效应良好等诸多优点,应用前景广泛[1-4],YPO4和LuPO4两种晶体的结构类似即都是正磷酸盐,且体系中阳离子是处于相邻周期的同一族元素.YPO4与独居石类似,具有熔点高,耐化学腐蚀,热稳定性良好等特性[5].有研究表明通过不同浓度的稀土元素对YPO4的参杂可以显著提高材料的荧光性能[6],晶体YPO4会随着掺入其中的稀土离子的浓度不同而表现出不同的闪烁性质[7,8].Lu-PO4与YPO4具有相似的晶体结构,在性质方面也有一定的相似性,即结构稳定、化学性质稳定、较高的熔点、耐腐蚀等特点[5].LuPO4在掺杂Nd3+离子后十分适合作为闪烁晶体材料,可以作为辐射探测器[9].也有研究建议可将LuPO4和YPO4作为固定放射性废物( 放射性稀土元素或三价锕系元素) 的基质和辐射监测传感器材料[10,11].
LuPO4与YPO4的优良性质吸引了许多科研人员的关注,Milligan 等人[12]在实验上通过三维单晶X 射线衍射的方法确定了LuPO4与YPO4的晶体结构; Wisniewski 等人[6]通过光谱分析研究了晶体缺陷对Nd 掺杂LuPO4和YPO4后发光性能的影响; Liu[13]团队通过第一性原理计算的方法研究了LuPO4晶体不同种氧空位的结构性质并且使用过渡态搜索计算探究了+2 价O 空位形成焦磷酸结构的机制.前人的研究多是将LuPO4与YPO4作为基质材料或是研究使用不同元素掺杂对晶体性能的改变,关于这两种晶体本身性质的理论对比讨论研究却少有报道.本文通过基于密度泛函理论( DFT) 量子力学第一性原理方法,利用CASTEP 程序模块[14,15]对两种磷酸盐晶体的电子结构进行模拟计算,具体将从能带结构、电子态密度、电荷密度、以及布局分析等方面展开.关于这两种晶体的电子结构的计算与分析可以辅助说明这两种材料的某些现象和机理,对于今后在实验上的补充以及实验内在机理的挖掘有一定的参考意义.
2 结构模型的建立和计算
图1 ( a) 、( b) 分别为LuPO4,YPO4的结构示意图,图中红色小球代表O 原子,紫色多面体代表P 原子,黄色多面体代表Lu 原子,蓝色多面体代表Y 原子.LuPO4,YPO4都属于四方晶系,空间群为I 41/amd,从结构上来看两种晶体具有一致性.LuPO4的晶格常数为: a=b=0.6783 nm,c=0.5947 nm[16]; YPO4的晶格常数为: a=b=0.6894 nm,c=0.6028 nm[16].
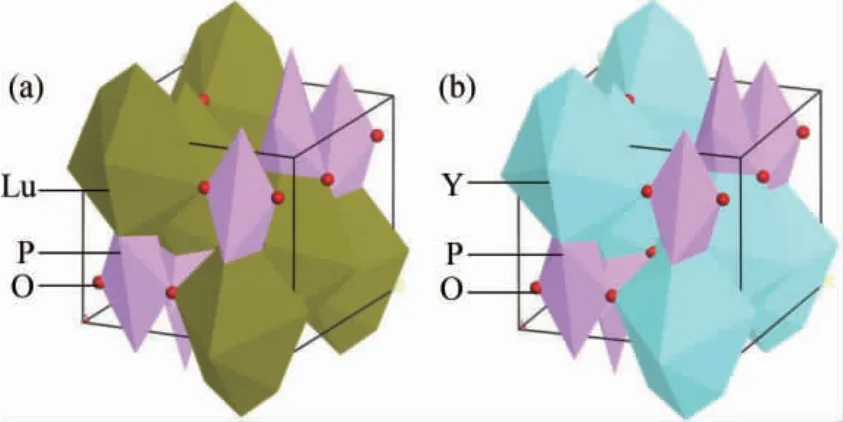
图1 ( a) LuPO4结构示意图( b) YPO4结构示意图Fig.1 ( a) Structural diagram of LuPO4,( b) structure diagram of YPO4.
首先对LuPO4体系,YPO4体系的几何结构进行优化.在此基础上对LuPO4体系和YPO4体系能量截断值都选取为400 eV,迭代收敛精度( SCF)均为1 ×10-6eV/atom,布里渊K 点网格均设置为4 ×4 ×4.离子实与价电子之间的相互作用采用超软赝势( ultra-soft pseudopotential,USP) 处理,关联泛函使用广义梯度近似( Generalized Gradient Approximation,GGA) 下 的 PBE ( Perdew Burke Ernzerhof) 泛函处理[17,18],体系电子波函数通过平面波基组展开.选取的价层电子组态分别为Lu: 4f145p65d16s2、Y: 4d15s2、P: 3s23p3、O:2s22p4.
3 计算结果分析与讨论
3.1 能带结构与电子态密度
费米能级附近的电子结构是影响固体物理性质的主要因素,选取费米能级附近的能带结构以及电子态密度进行绘图,如图2 所示,分别是两种晶体的能带结构( a) ( c) 与态密度图( b) ( d) ,图中虚线处是零点费米能级所在处,Z、A、M、Γ、R、X 为第一布里渊区的高度对称点.从图2( a) ( c) 中可以看出LuPO4价带顶( VBM) 与导带底( CBM) 位于同一高度对称点Γ 故属于直接带隙,对应的特征能量分别是EV=0 eV,EC=5.639 eV,故带隙为5.639 eV,实验上测得LuPO4带隙为8.6 eV[19]; YPO4价带顶和导带底均位于Γ 点也是直接带隙,特征能量分别是EV=0 eV,EC=4.884 eV,带隙为4.884 eV,实验值为9.2 eV[20].带隙的计算值相较于实验值均偏低,这在基于DFT 的计算中是普遍存在的,原因是密度泛函理论没有考虑交换关联势导数的不连续性[21],使得半导体和绝缘体的基态带隙被低估了,但同时Kohn-Sham 能带结构可以准确地预测禁带的中心,所以带隙的计算值比实验值小是正常的.较宽的禁带带隙使得两种晶体材料能够有效吸收高能射线,进而能够作为闪烁晶体材料或辐射探测器材料.
结合图2( b) ( d) 的态密度图可知LuPO4导带部分主要是由Lu 的5d 态电子贡献,费米能级附近的价带主要由Lu 的4f 态和O 的2p 态电子构成,其中O 的2p 态电子越过费米能级是价带顶的主要贡献者; YPO4导带部分主要是由Y 的4d态电子贡献,价带顶主要贡献也同样是来自于O的2p 态电子.两种晶体结构类似,相应的在费米能级附近态密度也类似,即导带底都是由金属原子的外层电子态贡献,价带顶都是由O 的2p 态电子贡献,这是由于阴离子和阳离子之间形成化学键后的杂化效应引起的[22].Lu 的4f 完全填充轨道导致LuPO4在费米能级左侧-2.5 eV 位置处出现一较尖锐的态密度峰,故与YPO4有所不同.在-7 eV 至-5 eV 范围内产生的两个态密度峰是由P 的3s 和3p 态电子所引起的.LuPO4在-24.5 eV 的深能级处的峰是由Lu 的5p 态电子贡献的.LuPO4和YPO4都在-17.4 eV、20.0 eV 处产生两个峰,分别是由O 的2s 态P 的3p 态、O 的2s 态P 的3s 态构成.
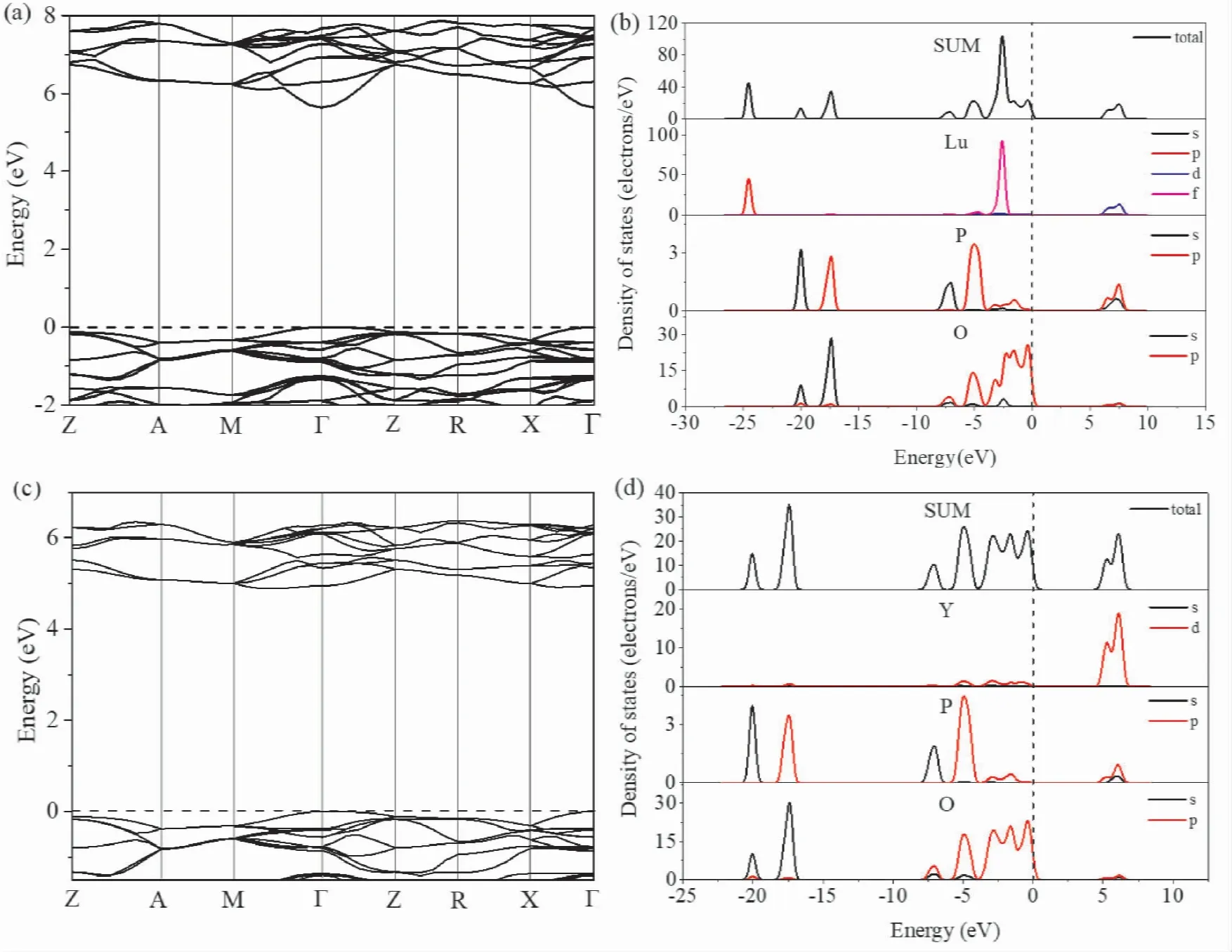
图2 能带结构: ( a) LuPO4( c) YPO4,态密度图: ( b) LuPO4( d) YPO4Fig.2 Band structures: ( a) LuPO4,( c) YPO4; densities of states: ( b) LuPO4,( d) YPO4.
3.2 电荷密度与布局分析
通过电荷密度图可以更加直观的观察晶体内原子价层电子的分布情况和相互作用.图3 是两种晶体的电荷密度图,其中红色小球代表O 原子,紫色小球代表P 原子,黄色小球和蓝色小球分别代表Lu 原子和Y 原子.从图中可以看出O原子附近聚集了大量的电子,呈现出较强的电负性,而P、Lu 和Y 作为失去电子的原子,其周围电荷密度就较低.进一步的定量分析则可以从表1 的Mulliken 原子布居数得到.对于LuPO4体系而言,其中O 原子得到0.97 e 的电子,P 原子和Lu原子则分别失去2.42 e 和1.47 e 的电子; 在YPO4体系中O 原子得到0.94 e 的电子,P 原子和Y 原子则分别失去2.18 e 和1.58 e 的电子.此外还可以从表中看出不同轨道电子的布局数,虽然布局分析产生的原子电荷的绝对大小几乎没有物理意义,因为它们对计算原子电荷的原子基函数表现出高度的敏感性[23].然而,考虑它们的相对值可以得到有用的信息,前提是使用一致的基函数进行计算[24].表2 给出了两体系中原子间的键长与重叠布局数,重叠布局数反映了两原子间成键电子的重叠程度,重叠布居数与键的共价性和键的强度有关.一般而言重叠布局数越小说明键的离子性越强,越大说明键的共价性越强.重叠程度越大则键长越短,键的强度也越强[24].在YPO4体系中就很好的反映了这种性质,O -P 键的重叠布局数(0.64) 大于O -Y 键的重叠布局数(0.31) ,相应的O-P 键的键长( 0.154 nm) 就小于O-Y 键的键长( 0.244 nm) ,即O -P 键的共价性较强而O -Y 键的离子性较强.在LuPO4体系中的情况也类似O -P 键的键长( 0.154 nm) 小于O-Lu 键的键长(0.235 nm) ,但值得注意的是O-Lu 键的重叠布局数( -5.75) 为负值,这是由于在Mulliken 布局分析中将基函数之间的耦合对轨道产生的贡献平分所造成的,这种平分的方式不尽然合理,尤其是在分析高能态占据轨道成分时负值的情况就经常出现[25].
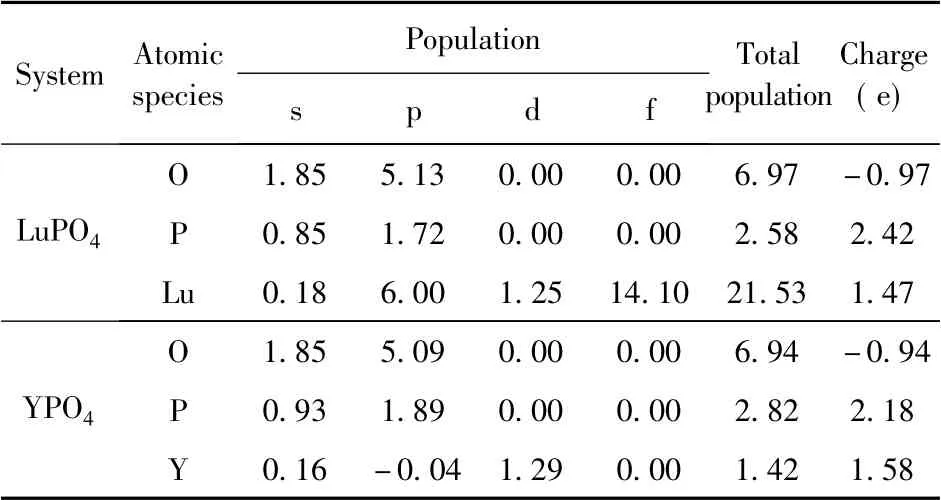
表1 原子布居数( Mulliken)Table 1 Atomic populations ( Mulliken)
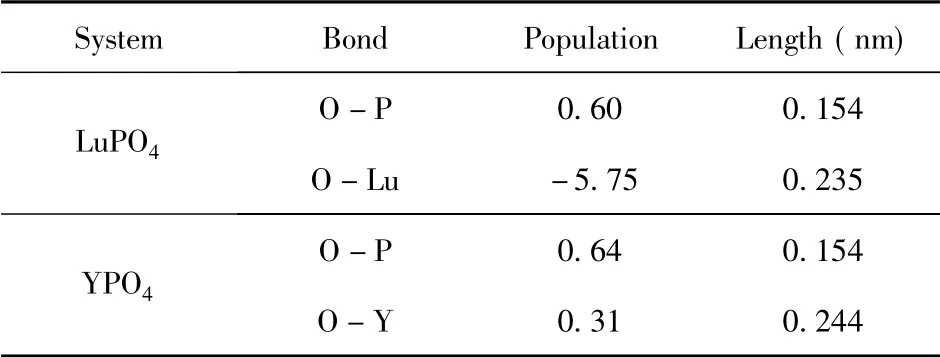
表2 LuPO4和YPO4的键长与重叠布局Table 2 Bond lengths and overlap populations of LuPO4 and YPO4
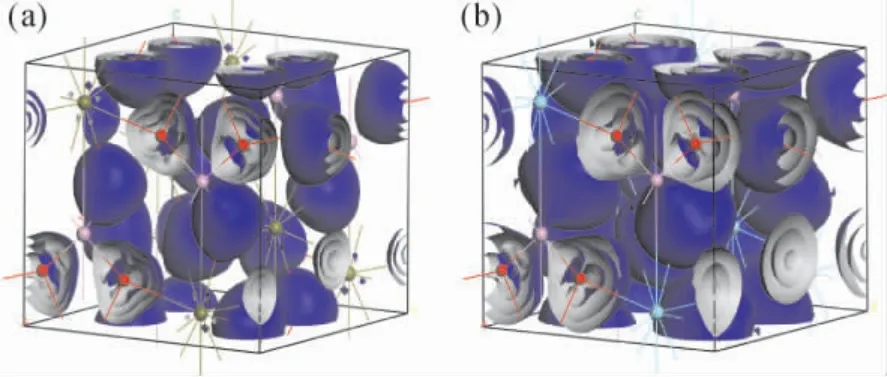
图3 电荷密度图: ( a) LuPO4,( b) YPO4Fig.3 Charge density diagram: ( a) LuPO4,( b)YPO4.
4 总 结
本文采用密度泛函理论( DFT) 第一性原理对LuPO4和YPO4两种磷酸盐晶体的电子结构进行计算模拟.结果表明: LuPO4的带隙为5.639 eV,YPO4的带隙为4.884 eV.通过对态密度的分析得知LuPO4的导带主要贡献来自Lu 的5d 态电子,费米能级附近价带主要由Lu 的4f 态和O 的2p 态电子贡献.YPO4的导带主要贡献来自Y 的4d 态电子,价带顶的主要贡献来自于O 的2p 态电子.通过电荷密度和布局分析得知了材料内部原子间的电荷转移情况,进而对原子间成键情况进行了分析与讨论,即O -P 键的共价性较强,O - Lu键和O-Y 键的离子性较强.
