Ni-C共掺杂AgSnO2触头材料电性能第一性原理计算
2022-03-04高振江
丁 璨,高振江,胡 兴,袁 召
(1.三峡大学 电气与新能源学院,宜昌 443002; 2.华中科技大学 强电磁工程与新技术国家重点实验室,武汉 430074)
1 引 言
机械式直流断路器是柔性直流电网的重要电气设备.触头是机械式直流断路器真空灭弧室的核心元件,起到接通、承载和分断电流的作用,断路器灭弧室中触头材料的导电、导热、抗熔焊和抗电弧侵蚀等性能直接影响到其稳定可靠性.现有的灭弧室触头[1]耐烧蚀强度还是不够,开断几次之后就会出现斑点裂痕,使得接触电阻增大,产生大量金属蒸汽,大量的金属蒸汽在电弧熄灭后很容易引起触头间隙的重燃从而开断失败,并使其寿命大大降低,亟需改进其导电和耐烧蚀等性能.触头材料作为触头性能变化的重要影响因素,它的优化选择和设计是机械式直流断路器设计极为重要的部分[2].
银-氧化物因其具有良好的导电性、抗熔焊性及接触电阻低等优点,被广泛应用为触头材料.1939 年Hensel 等学者开发出了AgCdO 电接触触头材料[3],又因其具有良好的灭弧性、抗电腐蚀性以及接触电阻低等优点,被称为万能触头.但是在使用过程中,会释放出有毒蒸汽Cd,之后取而代之的是AgSnO2触头材料,其具有较低的电阻率、优良的耐磨性和抗熔焊性而占据断路器触头材料的重要地位.该材料是以Ag 为主要成分,SnO2为掺杂材料,可以起到防止银融化后液体飞溅、增强银粘液度的作用.但是SnO2属于宽禁带半导体材料,禁带宽度约为3.6 eV,激子束缚能为130 meV[4],但是通过掺杂元素就可以改进材料的性能.近些年来许多学者在稀土元素、金属元素以及非金属元素掺杂SnO2改善光学、电学、热力学等性能[5-9]方面取得了一定的进展.赵彩甜等[10]针对La 掺杂AgSnO2触头材料的导电性能进行了第一性原理分析,发现La 掺杂比例为16.67%时导电性最佳; 彭彩云等[11]研究了不同浓度C 掺杂SnO2的第一性原理计算,发现随着掺杂浓度的增大禁带宽度减小,在光学方面,随着掺杂浓度的增大,体系向低能级方向移动幅度也越大;于淼等[12]研究了Mn-N 共掺杂SnO2电子结构的第一性原理计算,发现单掺杂和共掺杂均使得导电性增强,Mn-N 共掺杂使Mn 的掺入更加容易.
实验和理论上皆有对Ni 单掺杂和C 单掺杂SnO2的研究,但是对Ni-C 共掺杂AgSnO2触头材料电性能的理论分析还未见报道,因此本文运用第一性原理方法,计算了Ni-C 掺杂AgSnO2触头材料的能带结构、态密度和布局,从微观角度对机械式直流断路器真空灭弧室AgSnO2触头材料进行了优化分析,研究并对比了Ni、C 单掺杂和共掺杂SnO2材料的导电性,为改善直流断路器真空灭弧室AgSnO2触头材料导电性提供了一种思路.
2 计算方法与模型
2.1 计算方法
本实验采用CASTEP( Cambridge Sequential Total Energy Package) 软件来计算,在k 空间中,采用基于第一性原理平面波超软赝势来描述离子实与价电子之间的相互作用,倒格子空间中平面波截止能设置为400 eV,采用周期性边界条件,用广义梯度近似GGA 下的修正函数PBE 来处理电子与电子相互作用的交换关联能.参与计算的价电子为Sn 原子3d84s2、O 原子2s22p4、Ni 原子3d84s2、C 原子2s22p2.采用BFGS 优化算法对晶胞结构进行优化,收敛标准为: 体系总能量收敛值为2.0 ×10-6eV/atom,原子最大位移0.002 Å,原子间相互作用力0.05 eV/Å,晶体内应力偏差0.1 GPa,单原子能量2 ×10-5eV/atom,布里渊区积分采用Monkhors -Pack 形式,K 点设置为2×2 ×3.系统同时对计算体系进行结构优化.
2.2 晶体的结构及模型
电触头材料中的SnO2为金红石[13]型结构,空间群为136P4/MNM,属于体心四方晶系[14],每个本征SnO2原胞中包含2 个Sn 原子和4 个O原子,本研究使用Materials studio 软件建立了2 ×2 ×2 的SnO2超晶胞模型,超晶胞中含有16 个Sn原子和32 个O 原子,本文研究Ni 和C 掺杂对AgSnO2导电性能的影响,只需改善SnO2的导电性即可[15,16],掺杂模型通过原子替代的方法,将超晶胞中的非金属位O 用一个C 原子替代,金属位Sn 用一个Ni 原子替代,单掺杂和共掺杂的具体位置如图1 所示.
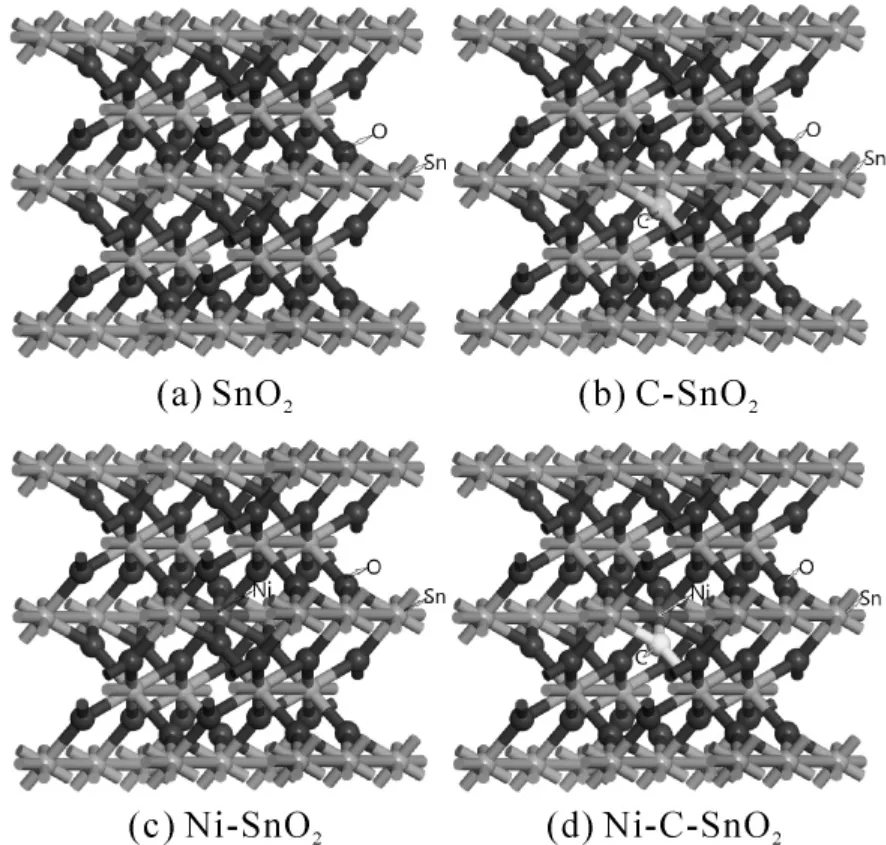
图1 超晶胞模型Fig.1 Supercell models
2.3 几何结构优化
几何优化后的超晶胞参数如表1,可以看出本征SnO2优化后的晶格常数为a=b=4.476 Å 和c=3.174 Å,与实验值[9]a=b=4.737 Å 和c=3.186 Å,α=β=γ=90°差距较小,说明本研究计算方法合理,这是由于采用了不同的近似算法导致的结果差异,但并不影响对结果的分析.掺杂后的晶格常数对于本征SnO2有不同程度的增大,晶胞体积也变大,由于Ni 原子半径(1.25 Å) 小于Sn 原子半径(1.58 Å) ,晶胞体积根据量子化学理论应该减小,但计算结果显示晶胞体积变大,这是由于Ni 替代Sn 以后会产生多余的正电荷,这些正电荷之间会产生排斥力,故晶胞体积会变大.C 掺杂以后,C 的原子半径(8.6 Å) 大于O 的原子半径(6.6 Å) ,所以体积变大.共掺杂时的体积是介于Ni 单掺杂和C 单掺杂之间,是因为C 的掺杂抵消了一部分Ni 产生多余的正电荷,故使得体积介于两者之间.
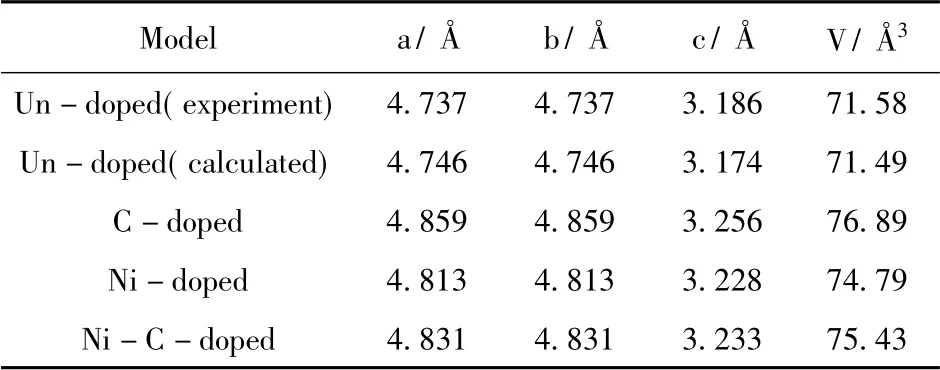
表1 SnO2(2 ×2 ×2) 掺杂前后的晶格常数与晶胞体积Table 1 Lattice constants and cell volumes of SnO2(2×2×2)before and after doping
3 结果与讨论
3.1 能带结构
本征SnO2、单掺杂和共掺杂后的能带结构图如图2 所示,本研究选取-5 ~10 eV 能带范围,能量零点为费米能级.图2( a) 为本征SnO2的能带图,带隙为1.064 eV 低于实验值3.6 eV,这是因为所用计算模块GGA 算法有一定的局限性,且在计算过程中低估了Sn 的5p 态与O 的2p 态之间的相互排斥作用,本文主要研究掺杂体系的对比值,所以并不影响本理论分析.能带图可以看出价带顶和导带底都在布里渊区高对称点G 处,可判定SnO2为直接带隙半导体材料.
Ni、C 单掺杂和Ni、C 共掺杂后如图2( b) ~( d) 所示,可以看出价带顶和导带底都在布里渊区高对称点G 处,说明掺杂后仍为直接带隙半导体材料,图2( b) 为C 掺杂SnO2的能带图,带隙为0.459 eV,同本征态相比禁带宽度明显减小,费米能级附近出现一条很窄的杂质能级,能带变得更加密集,体系能级数增多,这是因为C 原子最外层有4 个电子,C 原子替换O 原子后,会给体系提供更多的电子作为载流子,所以电子吸收更少的能量即可从价带顶通过杂质能级跃迁到导带底,从而增强了SnO2材料的导电性.图2( c) 为Ni 掺杂SnO2的能带图,带隙为0.746 eV,同本征态相比禁带宽度也是减小了,费米能级附近出现杂质能级,价带和导带均向费米能级移动,局域性增强.图2( d) 为Ni、C 共掺杂SnO2的能带图,带隙为0.283 eV,比单掺杂带隙更小,体系能带最为密集,价带和导带整体下移,费米能级附近出现更多的杂质能级,电子的离域性减弱,局域性更强,价带与导带之间出现电子跃迁的概率更大,金属导电性更强.

图2 单掺杂SnO2能带图( a) 本征SnO2; ( b) C 掺杂; ( c) Ni 掺杂; ( d) Ni-C 掺杂Fig.2 The band structures of( a) pure SnO2; ( b) C doped; ( c) Ni doped; ( d) Ni-C doped
3.2 电子态密度
本征SnO2、单掺杂和共掺杂后的总态密度和分态密度如图3 所示,由于远离费米能级的区域对材料性质影响不大[17-21],所以本研究取-25 ~26 eV 范围内的态密度.图3( a) 为本征SnO2的态密度图,可以看出在深部价带( -21 ~-14 eV)总态密度主要由Sn 的5s、5p 和O 的2s 轨道贡献,浅部价带( -8.4 ~0 eV) 的总态密度主要由O 的2p 轨道和少量Sn 的5s、5p 态共同提供,在费米能级左侧及价带顶处出现一个尖峰,主要是由O的2p 态贡献,在导带区域(0 ~26 eV) ,态密度主要是由Sn 的5s、5p 杂化作用和微量O 的2p 轨道共同贡献.
Ni、C 单掺杂和Ni、C 共掺杂后的总分态密度如图3( b) ~( d) 所示,图3( b) 为C 单掺杂,在价带-10 eV 附近,C 的2s 轨道与微量Sn 的5s、5p 轨道杂化出了一个较小的峰值,拓宽了价带的宽度,在费米能级两侧被C 的2p 轨道占据,也是能带图中出现杂质能级的来源,在价带( -8.4 ~0 eV) 的总态密度主要由O 的2p 轨道、C 的2p 轨道和少量Sn 的5s、5p 轨道形成,在导带区域(0 ~24.8 eV) ,态密度主要是由Sn 的5s、5p轨道、O 的2p 轨道和C 的2p 轨道杂化形成.图3( c) 为Ni 单掺杂的态密度图,在深部价带( -21.5 ~-15 eV) 处,总态密度由O 的2s 态和少量Sn 的5s、5p、Ni 的3d、4s 态电子贡献,在高能价带( -8.2 ~0 eV) 处的总态密度主要由Sn 的5s、5p、Ni 的3d 和O 的2p 轨道贡献,在导带部分(0 ~25.1 eV) 总态密度由Sn 的5s、5p、Ni 的3d、4s 和O 的2p 轨道贡献,在费米能级两侧被Ni 的3d 态电子占据,是杂质能级的来源.图3( d) 为Ni、C 共掺杂的态密度图,浅部价带和整个导带和单掺杂时类似,是由Sn 的5s、5p、Ni的3d、C 的2p 和O 的2p 电子价态贡献,Ni 的分态密度相比于Ni 单掺杂更加弥散,Ni -C 共掺杂也会使得Ni 更容易掺杂,导带底形成更多杂化峰,共掺杂时费米能级附近的峰值有所减小,局域性降低,产生了新的杂质能级,禁带宽度进一步减小,也增加了原子间的成键结合力,使得共掺杂后的导电性进一步增强,这与能带分析的结果相同.
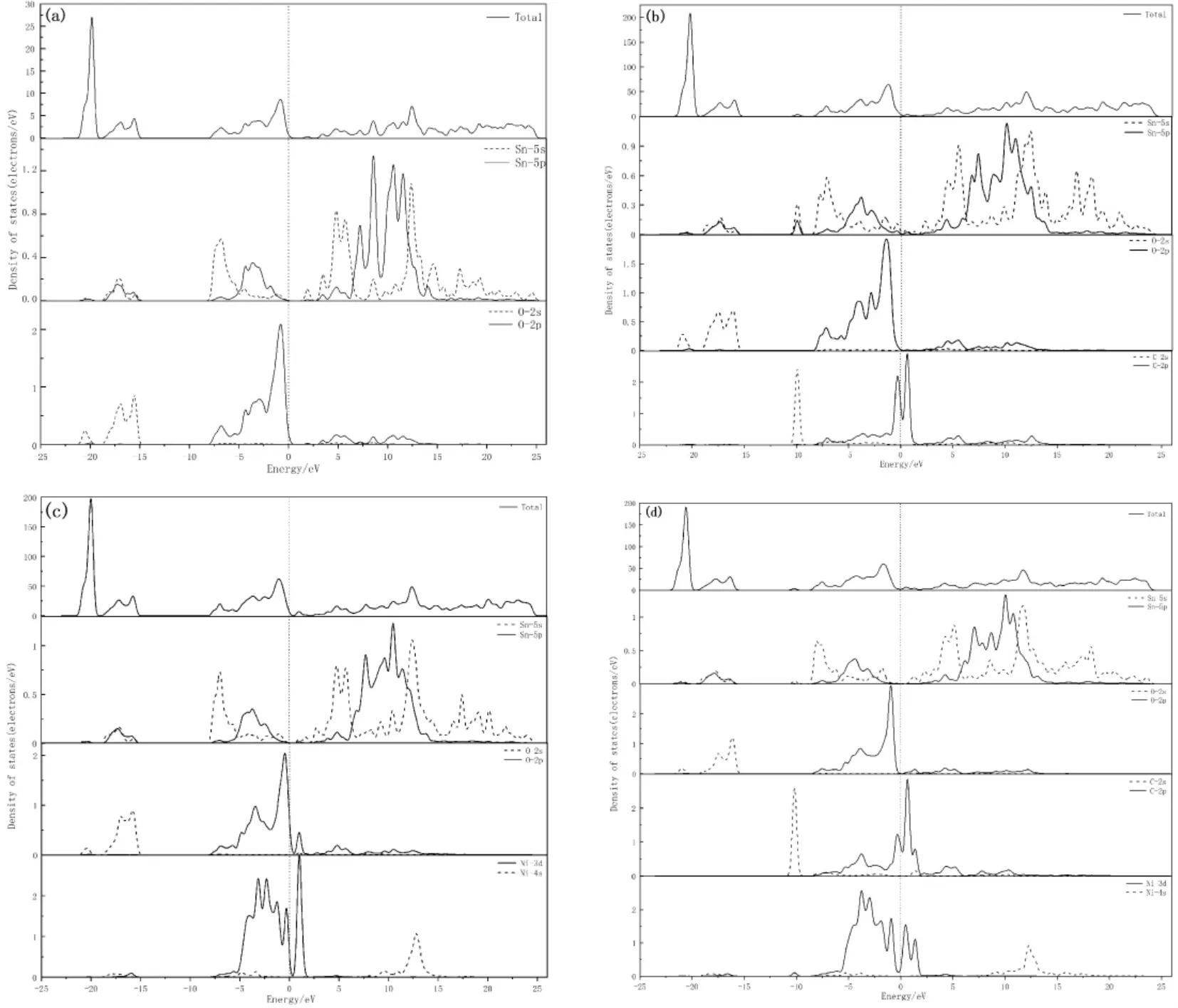
图3 态密度: ( a) SnO2; ( b) C 掺杂; ( c) Ni 掺杂; ( d) Ni-C 掺杂Fig.3 Densities of states: ( a) the pure SnO2; ( b) C doped; ( d) Ni-C doped
3.3 布局分析
表2 和表3 是根据仿真出来的结果文件得到的本征SnO2和Ni、C 单掺杂与共掺杂的各原子的电荷布局数和键布局数,所有数据为计算得到的平均值.
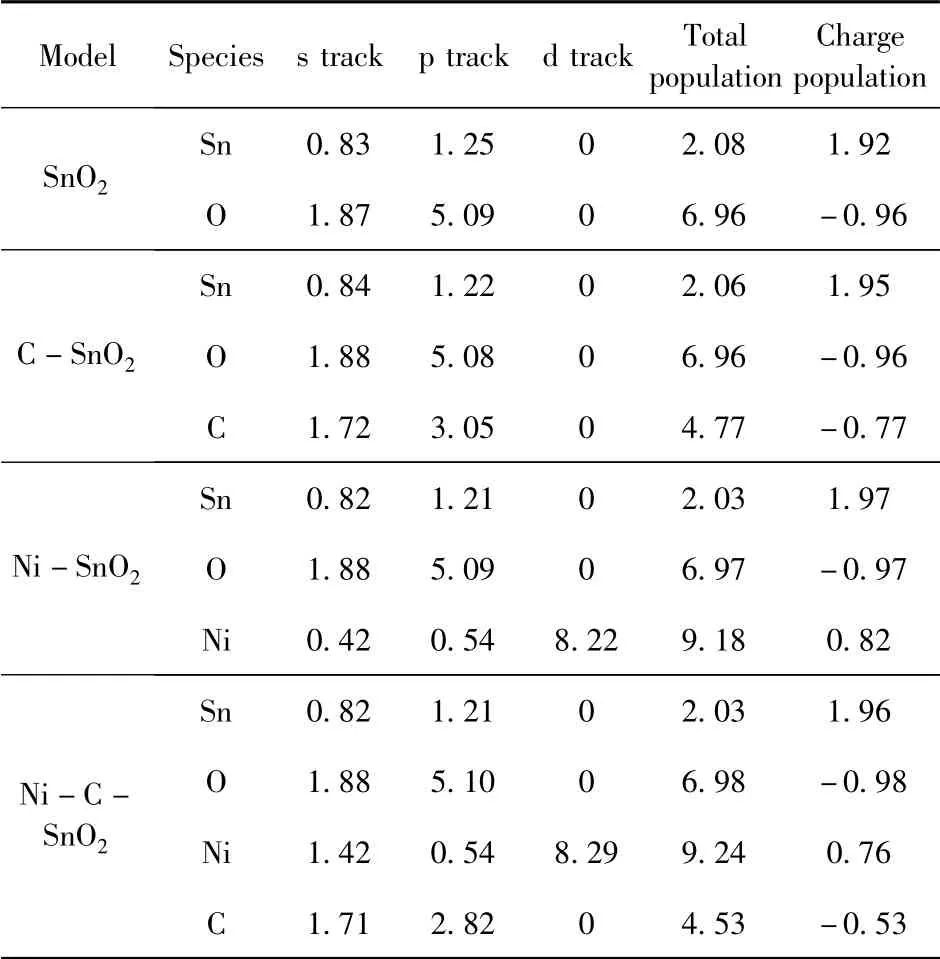
表2 电荷布局Table 2 Charge populations
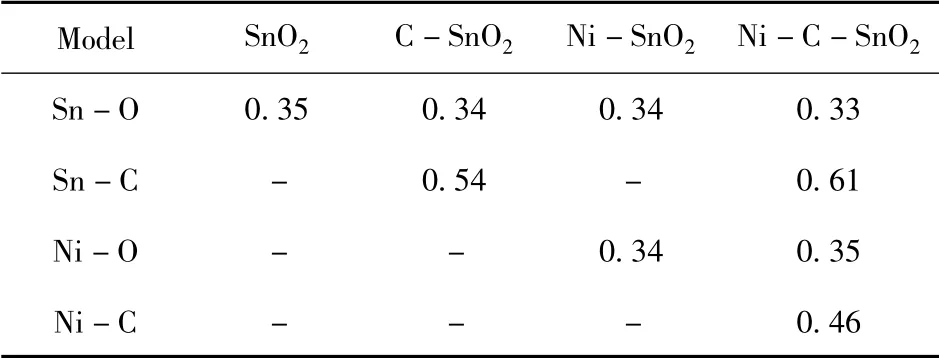
表3 键布局Table 3 Bond populations
由表2 中电荷布局可知,对于本征态SnO2,Sn 原子和O 原子的布局数分别为1.92 e 和-0.96 e,说明Sn 带正电失电子为主,O 带负电得电子为主,所以Sn 和O 之间形成离子键,O 的2p 轨道电子贡献值最大,与态密度分析一致.由Ni、C 单掺杂后的电荷布局数可知,C 的2p 轨道和Ni 的3d 轨道贡献最多的电子,C 的布局数为-0.77 e,得到电子显负电,Ni 的布局数为0.82 e,失去电子显正电,由于材料的得失电子,所以单掺杂可以增强导电性.Ni、C 共掺杂时,同样C 的2p 轨道和Ni 的3d 轨道贡献最多的电子,Ni 的布局数为0.76 e,C 的布局数为-0.53 e,同单掺杂相比绝对值有所下降,说明Ni 失电子数减小,Ni-O 键的离子性减弱,共价性增强,Sn 失电子数增大,Sn -O 键结合强度大于未掺杂,但共掺杂后的得失电子总数是多于单掺杂,也就有利于电子交换,导电性进一步提升,这与能带和态密度分析结果吻合.
由表3 的键布局数可知,本征SnO2中的Sn-O 键重叠布局数为0.35,Sn 和O 原子之间具有较强的共价性,存在强电荷密度重叠区; 在Ni、C单掺杂体系中,掺杂原子Ni、C 与Sn 原子之间发生杂化耦合,导致Sn -O 键的重叠布局数减小,共价性减弱,Ni、C 原子的电负性小于Sn、O 原子,Ni-O 键和Sn -C 键的共价性就小于Sn -O键,相互作用减小,Sn -C 键的布局数大于Ni -O 键,共价性较强; Ni、C 共掺杂后,Ni 原子和C 原子自由电子数重新分布,相互作用形成Ni -C 共价键,电子的共有化程度也进一步提高,金属性增强,导电性能进一步提升.
4 结 论
本研究基于密度泛函理论的第一性原理分析方法,采用CASTEP 软件中广义梯度近似GGA 的PBE 泛函来计算,得出了Ni、C 单掺杂和Ni -C共掺杂SnO2能带结构和电子态密度.结果如下:
1) 单掺杂和共掺杂均使得晶胞体积略微增大.
2) 单掺杂和共掺杂均使得禁带减小,且仍属于直接带隙半导体,在价带顶和导带底产生杂质能级,其中Ni-C 共掺杂时禁带最小,杂质能级最多,电子跃迁需要的能量更小,导电性最好.
3) 共掺杂时费米能级附近的峰值有所减小,局域性降低,原子间的成键结合力更强,也使得SnO2材料更加稳定.
4) 从布局分析可知,共掺杂后得失电子数增多,有利于电子交换,成键共价性增强,结构稳定性提高,导电性能提高.
5) 本文通过改善SnO2的性能来间接改善了AgSnO2触头材料的导电性,Ni -C 共掺杂SnO2时导电性最好,为改善机械式直流断路器真空灭弧室AgSnO2触头材料的电性能研究提供了参考.
