抗黄曲霉毒素B1多价纳米抗体融合蛋白的构建及活性分析
2022-03-04帅文苑何庆华钟引凤黄云祥刘传勇张乐平
帅文苑,何庆华,钟引凤,黄云祥,张 航,刘传勇,张乐平,涂 追*
(1.南昌大学 食品科学与技术国家重点实验室,江西 南昌 330047;2.南昌大学 食品学院,江西 南昌 330047;3.南昌大学 生命科学学院,江西 南昌 330031;4.江西省现代分析科学重点实验室,江西 南昌 330031)
黄曲霉毒素B1(AFB1)是由黄曲霉和寄生曲霉产生的有毒次级代谢产物[1-2],主要存在于发霉的花生、玉米、大米等农产品中[3-4],AFB1具有致癌性、致突变性、致毒性等,已被世界卫生组织的癌症机构划定为Ⅰ类致癌物。目前,检测农作物中AFB1的方法包括高效液相色谱法(HPLC)[5-6]、免疫传感器法[7]、免疫层析法(ICA)[8-9]、酶联免疫吸附法(ELISA)[10-11]等。免疫学检测方法基于抗体与抗原特异性结合,具有简便、高特异性、高灵敏度及高通量等优点。现有的AFB1免疫学检测方法采用的抗体主要有单克隆抗体、单链抗体等,然而单克隆抗体的制备较为繁琐,细胞培养及储存环境要求严格;单链抗体表达量低且常以包涵体的形式存在,应用受到一定的限制[12]。纳米抗体是通过克隆重链抗体的可变区并表达得到由一个重链抗体可变区构成的单域抗体(Variable domain of heavy chain of heavy chain antibody,VHH)[13],具有与原重链抗体相当的结构稳定性以及抗原结合活性,是目前已知的可结合目标抗原的最小单位,具有水溶性高,温度及pH耐受性好等优点。纳米抗体由于结构简单,适合于原核(如大肠杆菌)和真核表达系统进行高效表达,易于获得,已广泛应用于医疗[14]、诊断[15]、免疫分析[16]等领域。
增加免疫学检测方法中识别元件对待检物的亲和活性是提高检测灵敏度的主要途径之一[17]。已有研究表明,相比于单价纳米抗体,多价抗体形式具有更高的表观亲和力,有望建立灵敏度更高的免疫学检测方法[18-19]。本课题组前期从羊驼免疫库中筛选获得了针对黄曲霉毒素B1的纳米抗体(命名为G8),并将其应用于黄曲霉毒素的ELISA 检测以及样品前处理[20]。本研究以前期获得的抗AFB1纳米抗体G8为研究对象,利用纳米抗体分子量小,易于基因操作、原核表达的特点,通过串联融合表达的方法,构建了单价以及多价纳米抗体串联体,并分别与绿色荧光蛋白(GFP)编码片段融合,研究了多价化及融合表达对重组蛋白生物活性的影响,为后续建立基于荧光信号的免疫学检测方法奠定了基础,同时也可为优化免疫学检测中纳米抗体亲和活性提供了借鉴和思路。
1 实验部分
1.1 仪器与试剂
Varioskan LUX 多功能读数仪(美国Thermo 公司);ZHWY-2102C 恒温摇床(中科院武汉科学仪器厂);JY92-ⅡDN 超声波细胞粉碎机(宁波新艺超声设备有限公司);LAS 500 凝胶成像仪(BIO-RAD公司);DYY-8C 电泳仪(北京六一仪器厂);Nanodrop 1000 超微量分光光度计(美国Thermo 公司);DHG-9101·DSA 恒温培养箱(上海三发科学仪器有限公司);T100TMPCR 扩增仪(BIO-RAD 公司);NK-420 电热恒温水箱(常州诺基仪器有限公司);YLC-100 干式恒温器(江苏金怡仪器科技有限公司);Sorvall Stratos低温高速离心机(美国Thermo公司)。
含单价以及二价、三价抗AFB1纳米抗体的质粒pET30-G8、pET30-DiG8、pET30-TriG8,含绿色荧光蛋白的质粒PMCSG18hRahs-GFP,大肠杆菌DH5α、BL21(DE3)均由本实验室保存;人工抗原AFB1-BSA 由本实验室制备[21];氯化钠(NaCl)、磷酸氢二钠(Na2HPO4)、氯化钾(KCl)、磷酸二氢钾(KH2PO4)均为分析纯,购自上海阿拉丁生化科技公司;Lysis 平衡缓冲液(LE Buffer:50 mmol/L Na2HPO4,0.3 mol/L NaCl,pH 8.0);磷酸盐缓冲溶液(0.01 mmol/L PBS:137 mmol/L NaCl,2.7 mmol/L KCl,10 mmol/L Na2HPO4,2 mmol/L KH2PO4,pH 7.4);含0.05%吐温-20 的0.01 mmol/L 磷酸盐缓冲溶液(PBST);卡那霉素(Kana)、异丙基-β-D硫代吡喃半乳糖苷(IPTG)购自索莱宝公司;限制性内切酶SfiⅠ、NotⅠ、NdeⅠ,T4DNA 连接酶(TakaRa 公司);HRP 标记His 标签鼠单克隆抗体(Proteintech)、Ni2+-NTA 亲和层析(金斯瑞生物科技);DNA 片段纯化试剂盒(MiniBEST DNA Fragment Purification Kit Ver.4.0)(货号9716)、琼脂糖凝胶回收试剂盒(MiniBEST Agarose Gel DNA Extraction Kit Ver.4.0)(货号9762)(TakaRa 公司);质粒产物小提试剂盒(DP103-02,天根公司);经PAGE 纯化的引物由金斯瑞生物科技公司合成(见表1)。

表1 引物列表Table 1 List of primers
1.2 实验方法
1.2.1 原核表达载体的构建原核表达载体pET30-G8-GFP、pET30-DiG8-GFP、pET30-TriG8-GFP 的构建方法如图1 所示。经引物(G8-F0、G8-R0)获得G8 编码片段,限制性内切酶NdeⅠ和SfiⅠ将编码G8的基因克隆至载体pET30以获得载体pET30-G8。DiG8、TriG8通过含有编码柔性linker(GGSGG)的引物(G8-F1、G8-R1)及引物G8-F0、G8-R0 进行重叠延伸PCR(overlap PCR)而得,同样经由限制性内切酶NdeⅠ和SfiⅠ构建得到载体pET30-DiG8、pET30-TriG8。pET30-G8、pET30-DiG8、pET30-TriG8和由引物(F0、R0)扩增得到的GFP编码片段,用SfiⅠ和NotⅠ分别进行双酶切,试剂盒回收上述酶切片段。将纯化得到的载体与目的片段按1∶3的摩尔比用T4DNA连接酶于16 ℃孵育过夜,连接产物电转化大肠杆菌DH5α 感受态细胞,涂布于含50 μg/mL 卡那霉素的LB(LBKana)平板,37 ℃培养12 h。采用引物T7 prom和T7 term对单克隆进行菌落PCR验证,阳性克隆提取质粒后再进行双酶切验证,双酶切与预期相符的克隆送至南京金斯瑞生物科技公司测序。

图1 抗AFB1纳米抗体融合蛋白表达载体构建示意图Fig.1 Schematic of construction of anti-aflatoxin B1 nanobody fusion protein expression vector
1.2.2 纳米抗体的融合表达及纯化经测序验证结果正确的重组表达载体pET30-G8-GFP、pET30-DiG8-GFP、pET30-TriG8-GFP,电转化大肠杆菌BL21(DE3)感受态细胞,涂布于含50 μg/mL卡那霉素的LB(LB-Kana)平板,37 ℃培养12 h;挑取平板中的单菌落接种于含卡那霉素的5 mL LB 液体培养基中,37 ℃恒温摇床220 r/min 培养12 h;上述菌液按1%的比例接种于1 L LB-Kana 液体培养基中,37 ℃恒温摇床220 r/min 培养至OD600值为0.6 ~ 0.8(约2~3 h);上述培养液中加入IPTG 至终浓度为0.5 mmol/L,18 ℃过夜诱导培养。将诱导培养物6 500 r/min离心15 min,弃上清,收集菌体;1/10体积的LE 缓冲液重悬菌体,冰浴中超声波破碎菌体至澄清,破碎后的菌体于6 500 r/min 条件下离心20 min,分别收集破碎上清和沉淀。采用Ni2+-NTA 亲和层析纯化融合蛋白,SDS-PAGE 分析蛋白表达及纯化情况。
1.2.3 未融合、单价及多价纳米抗体的活性分析采用间接ELISA 分析重组蛋白与人工抗原的结合活性。将溶于PBS(pH 7.4)的人工抗原AFB1-BSA 以100 μL/孔加至酶标孔中,4 ℃包被过夜;PBST 洗板3次,加入5%脱脂牛奶,300 μL/孔,37 ℃封闭2 h;PBST 洗板3次,加入100 μL/孔重组蛋白G8(未融合、单价及多价),37 ℃孵育1 h;PBST 洗板3 次,加入100 μL/孔HRP 标记His 标签鼠单克隆抗体,37 ℃孵育1 h;PBST 洗板3 次,加入100 μL/孔TMB 底物显色液,37 ℃孵育10 min;加入50 μL/孔2 mol/L H2SO4终止反应,于酶标仪中测定OD450值。间接竞争ELISA 与间接ELISA 步骤基本相同,但在加入重组蛋白G8 时,应同时加入50 μL/孔重组蛋白G8(未融合、单价及多价)及50 μL/孔AFB1标准品(200、100、50、25、12.5、6.25、3.125、1.56、0.78、0.39、0.20、0 ng/mL)进行竞争反应。
1.2.4 重组蛋白的荧光检测在酶标板中分别加入相同浓度重组蛋白(G8-GFP、DiG8-GFP、TriG8-GFP)200 μL,放入多功能读数仪中,于荧光光谱模式下先设定激发波长为472 nm,扫描490~650 nm范围内的发射光谱;再设定发射波长为507 nm,扫描350~490 nm范围内的激发光谱,绘制激发及发射光谱图,并分别记录其最大激发和发射波长,及对应荧光强度。
2 结果与讨论
2.1 原核表达载体的构建
采用通用引物分别进行菌落PCR,结果显示获得了符合预期大小的基因片段(图2A)。将菌落PCR阳性的克隆提取质粒,pET30-G8-GFP、pET30-DiG8-GFP和pET30-TriG8-GFP 三种重组质粒经SfiⅠ和NotⅠ双酶切验证后均出现两条带,酶切后的3种载体片段5 633 bp、6 026 bp、6 419 bp和GFP基因片段745 bp与预期相符(图2B),经DNA测序证明三种表达载体构建正确。

图2 菌落PCR验证及重组质粒的双酶切验证Fig.2 Identification of colony PCR and double digestion of recombinant plasmid by agarose gel electrophoresis
2.2 融合蛋白的表达与纯化
将三种重组质粒pET30-G8-GFP、pET30-DiG8-GFP、pET30-TriG8-GFP 电转化大肠杆菌表达菌株BL21(DE3),经终浓度为0.5 mmol/L IPTG于18 ℃条件诱导表达12 h,分别收集诱导前后的菌体。SDS-PAGE显示,三种蛋白均可在大肠杆菌BL21(DE3)中可溶性表达,且经Ni2+-NTA亲和层析纯化得到的蛋白G8-GFP、DiG8-GFP、TriG8-GFP 的条带位置与预期分子量42.87、57.24、70.57 kDa大小保持一致。经试剂盒定量,三种蛋白的表达量分别为6.0、7.5、4.0 mg/L,且本实验未进行表达条件的优化,通过优化表达条件如诱导温度、诱导时间、IPTG 浓度等有望进一步提高表达量[20]。三种蛋白在表达过程中,融合蛋白G8-GFP 与DiG8-GFP 可溶性表达较多,易于破碎;而融合蛋白TriG8-GFP 包涵体表达占多数,较难破碎,推测由于该融合蛋白的分子量较大,蛋白在折叠过程中易以聚集形式表达形成包涵体,可溶性表达低,由此可以推测多价纳米抗体串联数目较多时,较难获得可溶性蛋白。

图3 三种融合蛋白的SDS-PAGE分析Fig.3 SDS-PAGE analysis of expression and purifi⁃cation of three fusion proteins
2.3 单价及多价纳米抗体的活性分析
采用间接ELISA对三种融合蛋白的结合活性进行分析,结果显示三种融合蛋白(G8-GFP,DiG8-GFP,TriG8-GFP)均能特异性结合人工抗原AFB1-BSA,说明纳米抗体在多价化后仍然保持抗原识别活性(图4)。经方阵滴定,确定人工抗原AFB1-BSA的工作浓度为2.5 μg/mL,融合蛋白G8、G8-GFP、DiG8-GFP 以及TriG8-GFP 的最佳工作浓度分别为0.5、2.125、3.125、8.90 μg/mL。
以AFB1标准品进行间接竞争ELISA,结果表明,G8、G8-GFP 以及DiG8-GFP 建立间接竞争ELISA 的IC50分别为12.10、14.10、2.19 ng/mL,提示单价融合蛋白IC50与未融合蛋白相似,双价融合蛋白IC50较未融合蛋白提高了5.5倍(图5)。三价融合蛋白TriG8-GFP的灵敏度降低,与标准品竞争趋势不明显,未能计算得到IC50值(图5),该结果与本实验室前期工作相吻合[22],推测可能是该融合方式对蛋白表达与折叠存在一定的影响。He 等[23]通过柔性连接子Linker(G4S)3共15个氨基酸构建多价纳米抗体,将其生物素化并固定在链霉亲和素衍生的细菌磁性纳米颗粒上,用于检测四溴双酚A,结果显示多价纳米体的结合活性由高到低依次为:三价、二价、一价。本研究构建的多价纳米体同样采用了柔性Linker,然而长度仅为前者的三分之一,提示纳米抗体的连接方式对活性有较大的影响。融合GFP蛋白的ELISA 数据偏差较未融合GFP 更大,且背景吸附值较高(图4、5),ELISA 的条件有待进一步优化以降低背景吸附和提高精密度。TriG8-GFP 由4 个串联的结构域组成,在表达过程中可能未完全正确折叠,导致非特异性吸附而不能被标准品阻断。

图4 间接ELISA分析3种融合蛋白的活性Fig.4 Bioactivity analysis of three fusion proteins by indirect ELISA
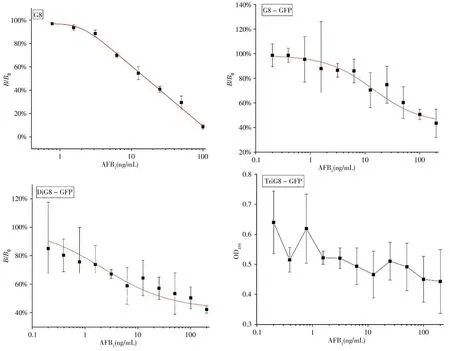
图5 间接竞争ELISA分析G8、G8-GFP、DiG8-GFP及TriG8-GFP的活性Fig.5 Indirect competitive ELISA of bioactivity analysis of G8,G8-GFP,DiG8-GFP and TriG8-GFP
2.4 重组蛋白的荧光检测
同一浓度(0.70 mg/mL)三种融合蛋白(G8-GFP、DiG8-GFP、TriG8-GFP)的荧光光谱如图6 所示。三种融合蛋白的最大激发波长均在472 nm左右,最大发射波长均在507 nm左右,与GFP相比最大激发和发射波长未发生改变。融合蛋白G8-GFP 与DiG8-GFP 的荧光强度均在3 000 及以上,与GFP相当且DiG8-GFP 表现出更高的荧光强度,而融合蛋白TriG8-GFP 的荧光强度仅在1 700 左右,相对荧光强度最弱(图6)。在表达过程中,推测三价串联的纳米抗体的融合蛋白TriG8-GFP的分子量较大,纳米抗体与荧光蛋白在表达过程中未能正确折叠,使得其荧光强度降低。由此推测,三价纳米抗体串联与荧光蛋白融合表达可能会影响其荧光活性,从而对基于荧光活性的免疫学检测存在一定干扰。
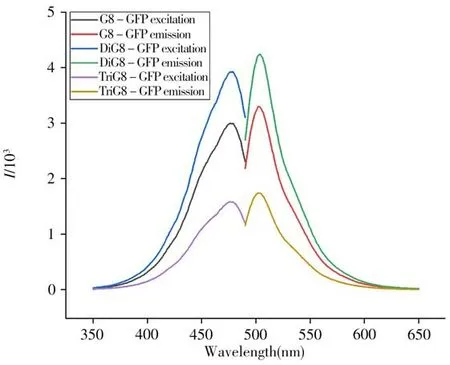
图6 三种融合蛋白的激发及发射光谱图Fig.6 Excitation and emission spectra of three fusion proteins
3 结 论
纳米抗体因其特殊结构可在大肠杆菌系统中进行高效可溶性表达,且串联后重组蛋白仍能可溶性表达。本研究以针对AFB1的纳米抗体和GFP 建立了研究模型,单价融合蛋白与未融合纳米抗体G8 相比结合活性并未显示较大差异,而双价纳米抗体与重组蛋白G8相比IC50提高了5.5 倍,且融合后GFP 的荧光强度及最大激发和发射波长并未发生改变,同时二价融合蛋白荧光活性表现较高,表明通过串联方式获得的针对AFB1二价纳米抗体的结合活性和荧光活性均较好。纳米抗体多价化的方式主要包括聚集体形式、串联表达等。本研究采用纳米抗体直接串联多价化方式,获得的二价纳米抗体与荧光蛋白的融合活性较好,为后续建立荧光免疫检测方法奠定了基础,具有一定的应用前景。
