鉴别牛肠道病毒感染复合PCR方法的建立及初步应用
2022-03-04常晓冉王浴光杨茗葳米日古丽买吐送胡俊英王新平吉林大学动物医学学院吉林长春30062吉林大学人兽共患病研究所教育部人兽共患病研究重点实验室吉林长春30062
章 凡,常晓冉,王浴光,杨茗葳,米日古丽买吐送,董 坤,胡俊英,王新平,2* (.吉林大学 动物医学学院,吉林 长春 30062;2.吉林大学 人兽共患病研究所 教育部人兽共患病研究重点实验室,吉林 长春 30062)
肠道病毒(enterovirus,EV)为小核糖核酸病毒科肠道病毒属成员[1-2],其所引起的感染严重危害人类健康和畜牧业的发展。该病毒为无囊膜单股正链RNA病毒,病毒粒子大小为20~30 nm,具有典型的二十面体结构[3]。根据国际病毒分类委员会(ICTV)的最新分类,肠道病毒属分为12个肠道病毒种(A、B、C、D、E、F、G、H、I、J、K、L)与3个鼻病毒种(A、B、C)[4],其中E种(EV-E)和F种(EV-F)主要感染牛,G种主要感染猪, H、J种主要感染灵长类动物,I种主要感染单峰骆驼,K、L为新的肠道病毒种。
牛肠道病毒(bovine enterovirus,BEV)感染于1959年首次由MOLL等[5]报道,之后许多国家也先后报道有本病的发生与流行[6-11]。李英利等[12]于2011年报道从内蒙古腹泻犊牛体内分离到我国第1株EV-F,确定我国牛群存在EV-F感染。本实验室于2012年从吉林省长春市某牛场发病牛群中分离获得首株EV-E HY12毒株[1-2],进而确定国内牛群存在EV-E感染。由于EV-E和EV-F属于两个不同的肠道病毒种,两者间的核苷酸差异很大,属于不同的血清型/基因型,因而常规方法难以将其区别[12]。近年来研究发现,国内许多地区牛群存在EV-E或EV-F肠道病毒感染,且某些牛群存在不同肠道病毒种的混合感染[13-15]。虽然有关检测EV-E或EV-F的研究逐年增加,但鉴别EV-E和EV-F感染的方法尚未见有报道。因此,迫切需要建立一种快速鉴别EV-E和EV-F的方法。本研究根据GenBank收录的EV-E和EV-F的全基因组序列比对分析结果,合成了2对特异性引物,建立了鉴别EV-E和EV-F的复合PCR方法,并初步用于临床样品的检测。
1 材料与方法
1.1 病毒株和临床样品牛细小病毒、牛病毒性腹泻病毒、EV-E HY12毒株、EV-F SD-S67毒株等病毒由本实验室鉴定保存。待检牛粪便样本采自长春地区牛场,共计32份,样品按常规方法处理、保存。
1.2 主要试剂和仪器TRIZol试剂、反转录试剂盒、TaqDNA聚合酶、DL2000 DNA Marker均购自TaKaRa公司;DNA凝胶回收试剂盒购自天根生化科技有限公司; ETC811 基因扩增仪为北京东胜创新生物科技有限公司产品;EPS300 电泳仪、4600SF紫外成像仪为上海天能科技有限公司产品; NanoDrop2000紫外可见分光光度计购自美国Thermo Forma公司;D1524R高速冷冻离心机购自北京大龙兴创实验仪器有限公司。
1.3 引物设计与合成根据GenBank收录的EV-E(D00214、DQ092792、DQ092786、KU17-2420、JQ690748、KF748290)和EV-F(DQ0-92770、DQ092795、DQ092794、AY462106、LC038-188)的基因组序列,以Meg Align(DNAStar)软件进行序列分析,利用Premier 5.0软件设计、合成2对引物,预计扩增EV-E和EV-F的基因片段大小分别为604,288 bp。引物由生工生物科技有限公司合成,引物信息见表1。
1.4 EV-E和EV-F单一PCR方法的建立应用提取的EV-E和EV-F感染细胞的RNA,合成cDNA,并以合成的cDNA为模板,进行病毒基因片段的PCR扩增。PCR反应体系为25 μL,即于反应体系中加入2×rTaq酶12.5 μL,模板0.5 μL,上、下游引物至终浓度为10 μmol/L,最后以ddH2O补至25 μL。反应程序:95℃ 5 min;95℃ 30 s,51℃ 30 s,72℃ 30 s,共35个循环;最后72℃ 10 min。同时以未加模板作为阴性对照。在此基础上进行EV-E和EV-F单一PCR的特异性及敏感性试验,PCR扩增产物以1.0%琼脂糖凝胶进行电泳鉴定。
应用DNA凝胶回收试剂盒,纯化和回收目的产物,并将其克隆到pMD18-T载体中,构建重组质粒 pMD18-EV-E和pMD18-EV-F。鉴定出的阳性重组质粒序列测定后,以紫外分光光度计测定浓度,并计算其拷贝数,于-20℃保存备用。

表1 复合PCR引物信息
1.5 复合PCR检测方法的建立条件优化以单一PCR扩增的条件为参考,利用梯度PCR程序对复合PCR反应体系进行优化。以不同的退火温度(50~60℃)和反应体系(10~30 μL)等条件进行PCR扩增。
1.6 特异性试验应用优化后的复合PCR方法,对混合感染EV-E和EV-F病料的cDNA及实验室保存的阳性牛细小病毒、牛病毒性腹泻病毒的核酸进行检测。
1.7 敏感性试验以不同稀释浓度的阳性质粒为模板,进行PCR扩增,确定出PCR扩增特异性片段所需要的最低拷贝数,即PCR的敏感性。
1.8 重复性试验以混合的EV-E和EV-F的cDNA为模板,应用建立的PCR方法重复检测3次,确定该方法的可靠性和重复性。
1.9 应用性试验应用建立的复合PCR方法,检测吉林省长春地区疑似BEV感染的32份临床样本,并与ELISA检测结果进行比较,计算两者符合率。
2 结果
2.1 单一PCR扩增结果利用P1/P2和P3/P4引物分别对EV-E和EV-F阳性DNA模板进行PCR扩增,琼脂糖凝胶电泳后可检测到 604 bp(EV-E)和 288 bp(EV-F) 大小片段(图1,2)。特异性结果均仅能扩增出EV-E或EV-F。敏感性结果显示,扩增出的EV-E和EV-F最低拷贝数为 3.67×102,5.21×103拷贝/μL。序列测定结果显示,大小为 604,288 bp 的片段分别为EV-E和EV-F。

A.EV-E单一PCR扩增结果(1.阴性对照;2.EV-E 扩增产物);B.EV-E特异性试验(1.阴性对照;2.EV-E;3.BVDV);C.EV-E敏感性试验(1~10.3.67×109~0拷贝;11.阴性对照); M.DL2000 DNA Marker
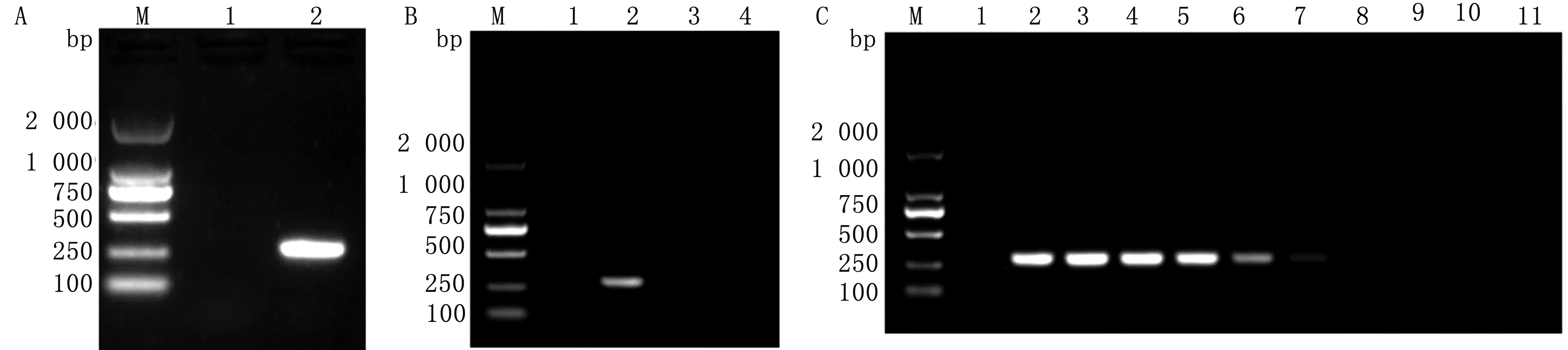
A.EV-F单一PCR扩增结果(1.阴性对照;2.EV-F扩增产物);B.EV-F特异性试验(1.阴性对照;2.EV-F;3.BVDV);C.EV-F敏感性试验(1~10.3.67×109~0拷贝;11.阴性对照);M.DL2000 DNA Marker
2.2 复合PCR的扩增及条件优化结果优化确定出的最终反应体系为10 μL,包括2×rTaq酶5 μL;引物P1、P2、P3、P4各为0.5 μL;模板0.5 μL;去离子水2.5 μL。最佳反应程序:95℃ 5 min; 95℃ 30 s,56℃ 30 s,72℃ 30 s ,共35个循环; 72℃ 10 min(图3,4)。以优化后的复合PCR的反应条件,将EV-E和EV-F的混合cDNA作为模板,进行PCR扩增,同时设阴性对照。扩增产物经琼脂糖凝胶电泳后,可见混合cDNA组同时扩增出2条带,其大小与单一cDNA组扩增条带相同(图5)。

M.DL2000 DNA Marker;1.阴性对照;2.5 μL;3.10 μL;4.15 μL;5.20 μL;6.25 μL;7.30 μL

M.DL2000 DNA Marker;1.阴性对照;2.51℃;3.52℃;4.53℃;5.54℃;6.55℃;7.56℃;8.57℃;9.58℃;10.59℃;11.60℃
2.3 复合PCR特异性结果应用复合PCR方法分别扩增EV-E和EV-F混合cDNA、牛细小病毒cDNA、牛病毒性腹泻病毒cDNA。除EV-E和EV-F混合cDNA组扩增出2条特异性条带,其余均未扩增出条带,表明复合PCR具有良好的特异性(图6)。
2.4 复合PCR的敏感性试验结果将紫外分光光度计测定的pMD18-EV-E和pMD18-EV-F质粒充分混匀后,确定出pMD18-EV-E和pMD18-EV-F的初始拷贝数约为 3.67×1010, 5.21×1010拷贝/μL。以10-1~10-10的梯度进行稀释,进行PCR扩增。建立的复合PCR方法检测EV-E和EV-F的最低检测滴度为 3.67×102,5.21×103拷贝/μL(图7)。
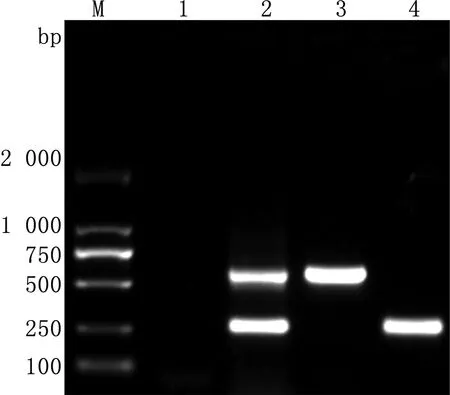
M.DL2000 DNA Marker;1.阴性对照;2.复合PCR扩增EV-E和EV-F混合cDNA产物;3.EV-E PCR产物;4.EV-F PCR产物
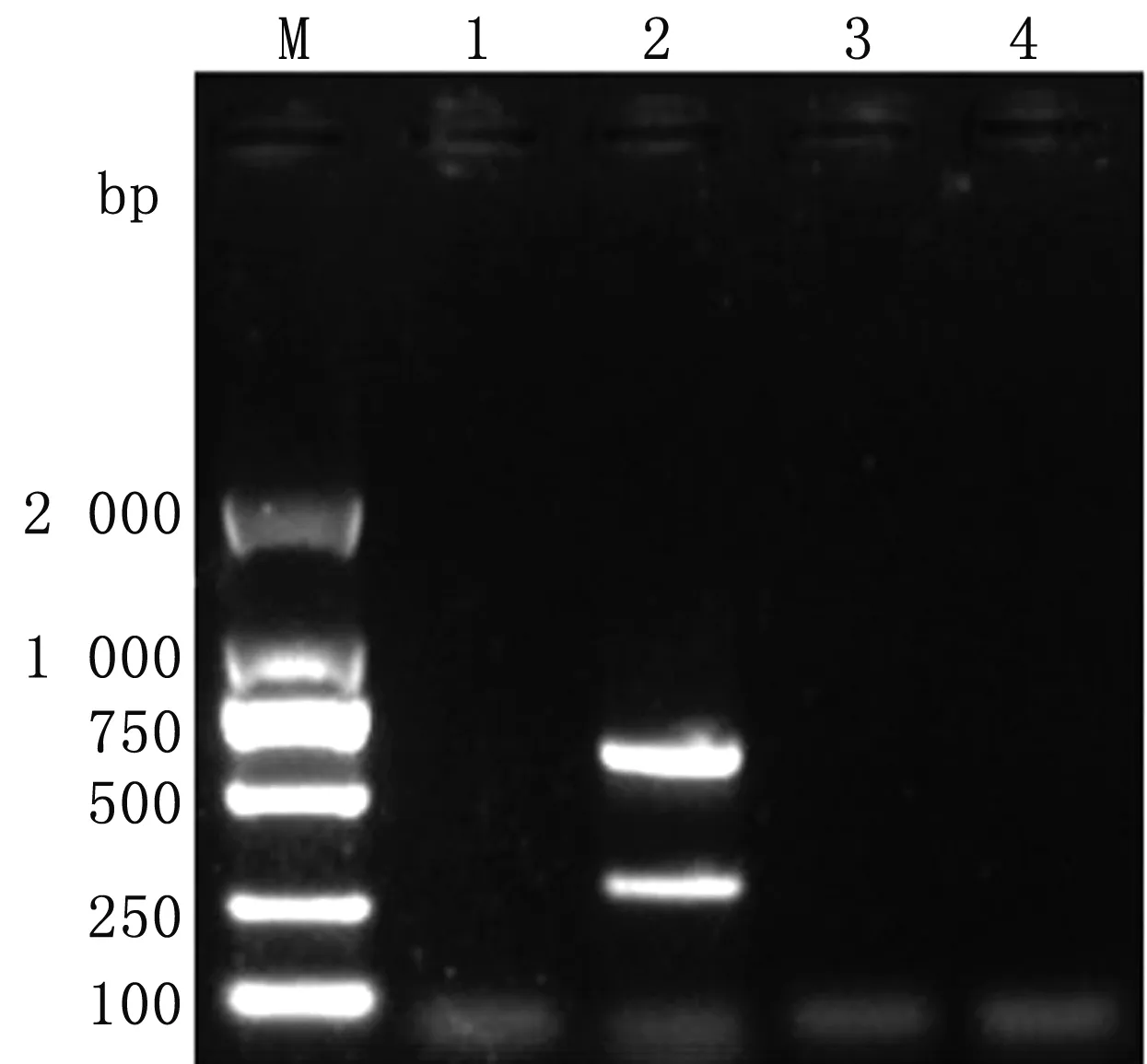
M.DL2000 DNA Marker;1.阴性对照;2.EV-E和EV-F混合cDNA扩增结果;3~4.复合PCR扩增牛细小病毒和牛病毒性腹泻病毒结果
2.5 复合PCR的重复性结果以复合PCR分别对3份样品进行3次重复性检测,结果EV-E和EV-F均为阳性,且阴性对照正常(图略),说明试验方法重复性良好。
2.6 复合PCR的初步应用结果用建立的复合PCR方法和ELISA方法对32份临床样品进行检测。所建立的复合PCR方法检测出的EV-E阳性数为9份(阳性率28.13%),EV-F阳性数为11份(阳性率34.38%),混合感染率为15.63%。与ELISA检测结果符合率为100.0%(图8,表2)。
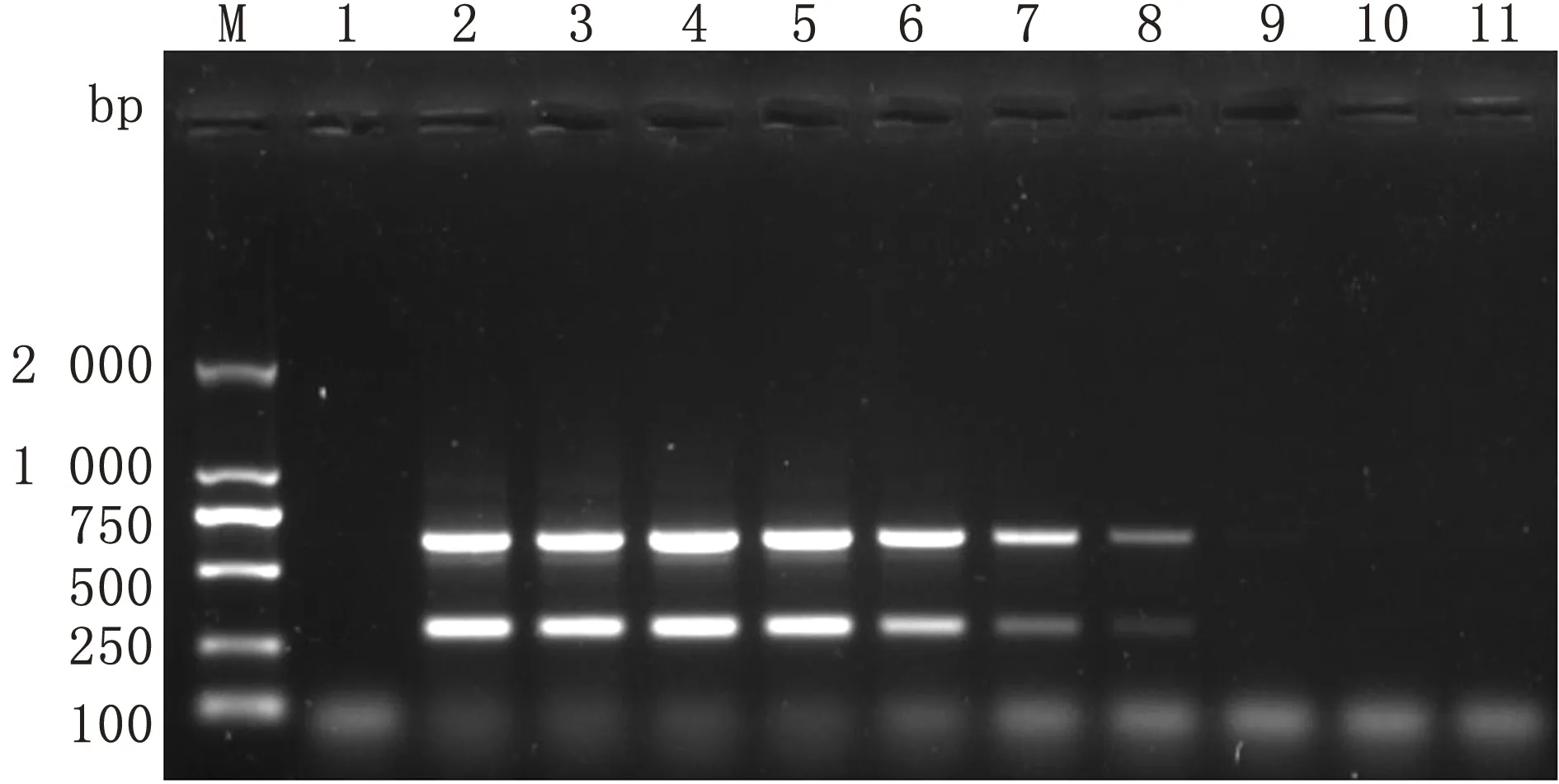
M.DL2000 DNA Marker;1.阴性对照;2~11.pMD18-EV-E和pMD18-EV-F混合质粒作10-1~10-10稀释后的复合PCR结果
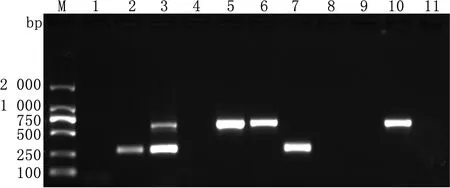
M.DL2000 DNA Marker;1.阴性对照;2~11.部分粪便样品的复合PCR结果
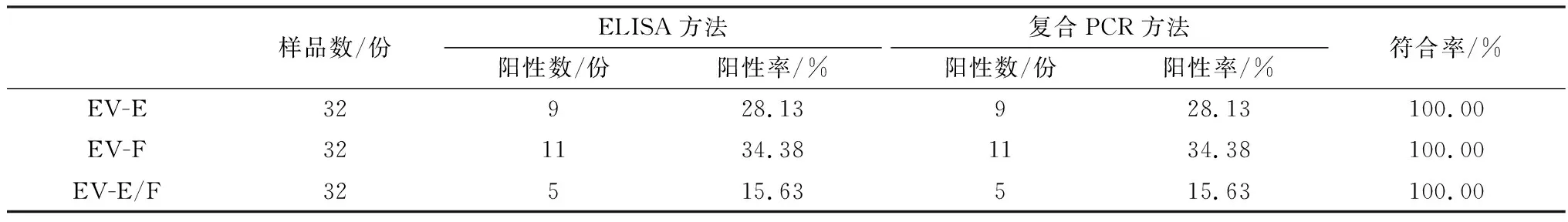
表2 RT-PCR和ELISA检测临床样品结果
3 讨论
BEV感染为国内新发的传染病,有关本病的研究近年来虽然取得了一些进展,但其诊断方法与流行病学研究仍然缺乏。目前,国内检测 BEV感染的方法主要有病毒分离、RT-PCR、实时荧光定量PCR、双抗体夹心ELISA等方法[16-18],这些方法虽然在确定国内牛群存在新发EV-E和EV-F感染方面发挥了重要作用,但由于它们只针对单一病毒血清型或基因型,因而在诊断 BEV感染中,难以揭示出牛群中的真实感染情况。本课题组前期对国内不同地区 BEV感染进行流行病学研究,发现国内许多地区的牛群同时存在EV-E和EV-F病毒混合感染的情况[13-15],导致 BEV感染的诊断与防控更加复杂。为了能够迅速同时确定出牛群中感染BEV的基因型或血清型,本研究基于对EV-E和EV-F的基因组的比对分析结果,分别设计了特异性扩增EV-E和EV-F的引物,并以此建立了可以依据扩增片段的大小进而鉴别EV-E和EV-F感染的复合PCR方法。特异性试验结果表明,建立的复合PCR仅能够检测出 BEV,具有较高的特异性;敏感性和重复性试验结果显示,该方法具有较高的敏感性和良好的重复性,且快速简便,可用于 BEV基因型/血清型的鉴别及诊断与流行病学调查研究。
本试验同时将2对引物的GC含量控制在50%~59%,以尽量降低GC含量差异对复合PCR的影响。在进行PCR反应体系及条件的优化中,发现应用25 μL体系进行复合PCR反应时,EV-E的扩增效率明显高于EV-F的扩增效率,导致EV-F检测结果呈现假阴性。而在10 μL反应体系下,两种引物扩增效率都比较高,从而使同时高效检测EV-E和EV-F成为可能。
