NLRP3炎症小体在年龄相关性黄斑变性中的作用
2022-02-23陈露综述谢平胡仔仲审校
陈露 综述 谢平 胡仔仲 审校
南京医科大学第一附属医院眼科 210029
陈露现在徐州医科大学附属徐州市第一人民医院眼科 221002
年龄相关性黄斑变性(age-related macular degeneration,AMD)是一种视网膜退行性疾病,表现为进行性视网膜色素上皮(retinal pigment epithelium,RPE)细胞功能失调,引起光感受器细胞死亡,最终导致视觉功能丧失[1]。早期AMD的病理特征为玻璃膜疣和RPE色素异常。晚期AMD有2种临床表现形式,一种是非新生血管性AMD,也称为萎缩性、非渗出性或干性AMD,其特征为延伸至黄斑中心凹的地图样萎缩(geographic atrophy,GA);另一种为新生血管性AMD,也称为渗出性或湿性AMD,其特征为脉络膜新生血管(choroidal neovascularization,CNV)的形成。流行病学研究表明,45岁以上人群中AMD的全球患病率为8.7%,我国50岁以上人口AMD发病率高达10.5%[2-3]。随着经济的发展和人口老龄化,预计2040年AMD患者人数将增加至2.88亿[2]。AMD严重影响患者的生活,并造成巨大的社会负担。然而目前尚无有效治愈AMD,尤其是干性AMD的方法[4],因此深入研究AMD的发病机制,探寻治疗的新方法已迫在眉睫。目前,AMD的病因和发病机制尚不清楚,研究表明遗传和环境等多种因素均可导致AMD的发生[5]。与AMD发展相关的炎症通路已逐渐成为近年来的研究热点[6-7]。本文就核苷酸结合寡聚化结构域样受体3(nucleotide-binding oligomerization domain like receptors 3,NLRP3)炎症小体及其下游因子与AMD的发病和治疗的关系进行综述,以期为AMD的治疗提供新的思路。
1 NLRP3炎症小体
1.1 NLRP3炎症小体的结构
核苷酸结合寡聚化结构域NOD样受体(NOD-like receptors,NLRs)是一组具有共同结构的细胞模式识别受体家族(pattern recognition receptor,PRRs),包括3个结构域,中央核苷酸结构域介导腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)依赖的自我寡聚化,其C-末端富含亮氨酸重复序列,用以识别配体,N-末端为可变的交互区域,负责与同型蛋白质的交互作用。PRRs可识别宿主来源的危险信号分子(damage-associated molecular patterns,DAMPs)和各种病原体编码的病原相关分子模式(pathogen-associated molecular patterns,PAMPs),并通过激活核转录因子(nuclear factor,NF)-κB、AP-1或IRF转录因子参与细胞内信号转导。作为NLRs成员之一,NLRP3炎症复合体包含NLRP3蛋白、caspase-1和含caspase募集结构域的凋亡相关颗粒样蛋白(apoptosis-associated speck-like protein containing a caspase recruitment domain,ASC)3个部分。当细胞受到各种危险信号刺激时,NLRP3炎症小体可通过ASC招募前体caspase-1并使其成熟,进而诱导白细胞介素(interleukin,IL)-1β和IL-18的成熟和分泌[8]。
1.2 NLRP3炎症小体的激活
研究表明所有PAMPs和DAMPs通过共同的途径激活炎症小体,但其机制尚未完全明确。NLRP3炎症小体可由多种刺激激活,如细胞死亡引起的细胞外ATP或透明质酸浓度增加、阿尔茨海默病相关的β-淀粉样蛋白表达增多、细胞外高糖、细胞外渗透压或pH改变等[9-12]。炎症小体激活过程的第一步为炎症小体启动,需要启动信号的刺激,由Toll样受体(Toll-like receptor,TLR)和NF-κB上调NLRP3、IL-1β和IL-18前体的RNA表达水平。而NLRP3寡聚化需要激活信号的刺激,胞外或胞内PAMPs和DAMPs刺激细胞装配NLRP3炎症复合体,最终使成熟的IL-1β和IL-18释放到胞外,此过程包括K+外流增加、Ca2+信号增强、溶酶体失稳定和破裂、活性氧产生等多种途径(图1)[13]。除了一般的细胞凋亡,NLRP3炎症小体同时引发细胞焦亡参与疾病的发生和发展[14-15]。
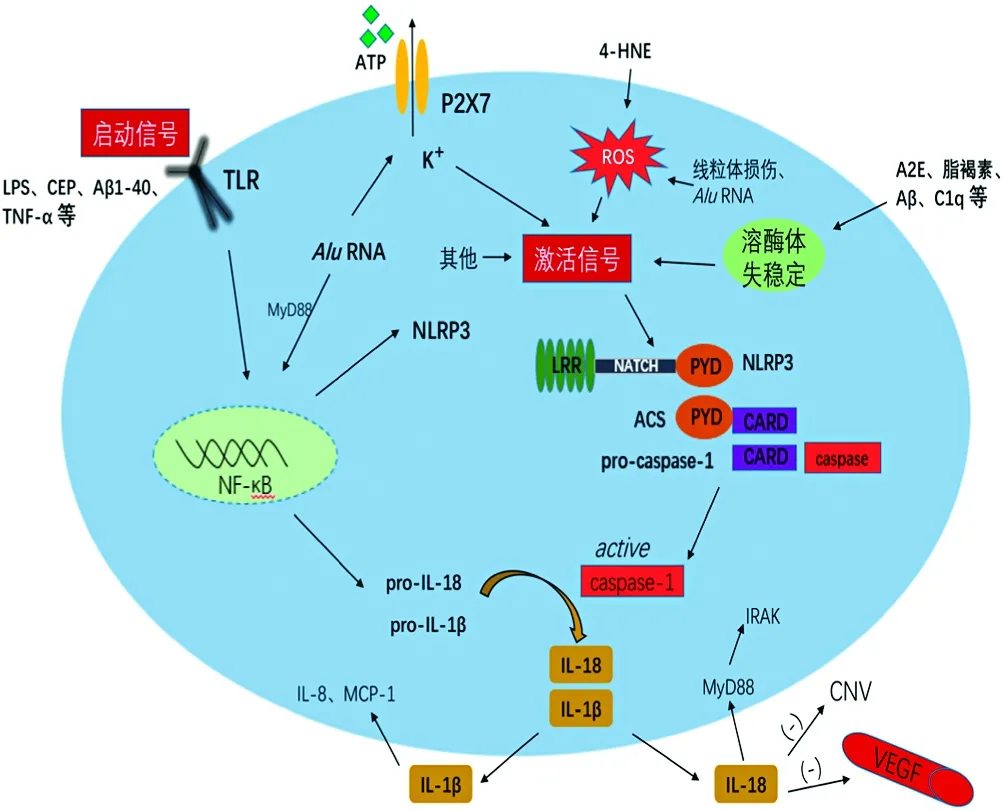
图1 NLRP3炎症小体在RPE细胞中的激活过程 启动信号通过刺激TLR和NF-κB上调NLRP3、IL-1β和IL-18前体的RNA表达水平,激活信号引起NLRP3寡聚化,装配NLRP3炎症复合体,最终使成熟的细胞因子IL-1β和IL-18释放
1.3 NLRP3炎症小体在疾病中的作用
NLRP3炎症小体及其下游信号分子已被证明参与多种疾病的病理过程。β-淀粉样蛋白是阿尔兹海默病患者老年斑的主要成分,其被证明可以激活小胶质细胞中NLRP3炎症小体,而NLRP3基因敲除小鼠较普通阿尔兹海默病模型小鼠运动、学习和记忆能力有所改善[16]。动脉粥样硬化相关危险因素均可诱导巨噬细胞内NLRP3炎症小体活化,促进IL-1β等炎性因子的释放,加重炎症反应[17]。此外,NLRP3炎症小体也参与2型糖尿病、呼吸道变态反应性疾病、缺血-再灌注疾病、肿瘤等的发生过程[18-19]。青光眼、糖尿病视网膜病变等眼科疾病也被证明与NLRP3炎症小体相关[20]。
2 NLRP3炎症小体与AMD
2.1 NLRP3炎症小体在眼底各细胞中的表达
NLRP3炎症小体可在RPE细胞、小胶质细胞、Müller细胞、血管内皮细胞等中表达[21]。而在AMD长期的病理过程中,小胶质细胞的活化与视网膜损伤密切相关,提示NLRP3炎症小体很有可能参与AMD的发生过程[22-24]。本研究团队通过建立视网膜光损伤模型发现,NLRP3炎症小体在视网膜小胶质细胞中被激活[25]。而活化的NLRP3炎症小体诱导产生的IL-1β、IL-18等细胞因子可能主导视网膜结构和功能损伤[26]。此外,通过NLRP3炎症小体抑制剂Mcc950抑制NLRP3炎症小体的激活可以缓解RPE细胞死亡及IL-1β分泌[27]。
2.2 NLRP3炎症小体参与AMD的发生
最初有研究者在GA患者的RPE细胞中发现大量NLRP3炎症小体、IL-18以及活化的caspase-1和髓样分化因子88(myeloid differentiation factor 88,MyD88),提出NLRP3炎症小体可能参与AMD的发生[28]。自此,NLRP3炎症小体逐渐成为AMD新的研究热点。AMD玻璃膜疣及其补体蛋白组分可以激活外周骨髓和单核细胞中的NLRP3炎症小体和caspase-1,进而诱导IL-1β和IL-18的分泌。Doyle等[29]通过羧乙基吡咯处理小鼠建立干性AMD病理学模型发现,视网膜巨噬细胞中存在活化的NLRP3炎症小体和caspase-1。RPE细胞的氧化应激也可引起NLRP3炎症小体及下游IL-18、IL-1β的mRNA表达升高[30-31]。此外,RPE细胞内溶酶体失稳定也会引发NLRP3炎症小体的激活,进而诱导炎性因子IL-1β的释放[32]。以上研究充分表明NLRP3炎症小体及其下游的产物可诱导AMD的进展,尤其是干性AMD。
DICER1缺失是引发AMD的另一重要原因。DICER1是一种微小RNA(microRNA,miRNA)加工酶,DICER1缺失可导致Alu RNA序列在RPE细胞中累积,引起视网膜GA改变。细胞中过量的Alu RNA可以激活NLRP3炎症小体,并通过非TLR途径激活MyD88产生细胞毒性,引起干性AMD表现[28]。抑制NF-κB及P2X7嘌呤受体可阻断此过程,P2X7嘌呤受体可以与高浓度ATP结合,打开离子通道,引起K+外流和细胞膜上半通道蛋白小孔形成,使胞外的配体进入细胞质,促进NLRP3炎症小体的激活[33]。NLRP3炎症小体的激活可以诱导IL-18释放,进而激活下游IL-8,造成RPE细胞损伤,导致AMD的发生。另一方面,DICER1缺失可引起下游miRNA调控异常,最终导致RPE细胞损伤[34]。
NLRP3炎症小体的激活受多种上游成分的调控,玻璃膜疣其他组分也可激活RPE细胞内的NLRP3炎症小体。β-淀粉样蛋白1-40可以上调RPE细胞中IL-6、TNF-α、IL-1β、IL-18、NLRP3炎症小体和caspase-1等炎症相关因子的mRNA表达。玻璃体内注射β-淀粉样蛋白1-40可引起玻璃体中IL-1β和IL-18浓度升高,这一过程可能与NADPH氧化酶和线粒体活性氧产生有关[12]。脂褐素成分A2E可以激活NLRP3炎症小体,诱导RPE细胞产生IL-1β等多种细胞因子和趋化因子,对小胶质细胞也具有激活作用。在ABCA4基因敲除小鼠RPE细胞中存在A2E累积,也可以观察到IL-1β表达水平升高[35]。
NLRP3炎症小体已被证明同时参与新生血管性和干性AMD的发生。血管内皮生长因子(vascular endothelial growth factor,VEGF)-α的增加可以激活NLRP3炎症小体,而NLRP3炎症小体缺乏可以减轻VEGF-α引起的CNV损伤和RPE屏障破坏[36]。随着研究的持续进展,线粒体DNA、7-酮基胆固醇、细胞外高浓度的氯化钠、氧化低密度脂蛋白等多种因素均证明可升高NLRP3炎症小体、caspase-1和下游IL-1β、IL-18蛋白的表达水平,参与AMD的发生[11,31,37-40]。线粒体DNA损伤-环化GMP-AMP合成酶(cyclic GMP-AMP synthase,cGAS)-caspase-4角度也同样证实了NLRP3炎症小体激活在干性AMD中的作用机制[41-42]。本研究团队的研究表明,慢性蓝光照射可以活化视网膜小胶质细胞,通过胞内NLRP3炎症小体的组装和IL-1β的分泌引起感光细胞死亡,导致光诱导的视网膜病变,引发AMD,而趋化因子CCR2基因敲除可使小胶质细胞胞质内活化的NLRP3炎症小体及其下游因子表达减少,视网膜光损伤缓解[25]。
3 NLRP3炎症小体下游因子IL-18和IL-1β与AMD
3.1 IL-18和IL-1β的生理作用
IL-1β和IL-18作为IL-1细胞因子家族成员,它们首先被合成为无活性前体,需要经过caspase-1裂解才可以产生成熟的、具有生物活性的细胞因子。IL-1β作为一种经典的炎症激活剂和调节剂,其异常表达与一系列自身免疫性疾病有关,阻断IL-1β信号可以起到一定的治疗作用。IL-18的作用具有两面性,在一定的微环境中,IL-18可以响应辅助性T细胞的刺激,被证明与多种炎症状态有关[28]。然而另有研究显示,IL-18在AMD中发挥保护作用[29]。这表明在细胞不同的免疫状态或炎症的不同过程中IL-18既可以发挥保护作用,又可以发挥促炎作用。
3.2 IL-18和IL-1β在AMD中的作用
NF-κB和MAPK通路激活产生的IL-1β可以诱导RPE细胞中IL-8和单核细胞趋化蛋白1的表达和分泌[43]。IL-1β长期慢性干预可以增加RPE层的通透性,影响紧密连接蛋白的表达[44]。阻断IL-1β信号转导可以显著减少VEGF-A诱导的AMD模型小鼠中CNV病变的数目[36],给予IL-1受体拮抗剂anakinra同样可以降低模型大鼠中激光诱导CNV的面积[45]。IL-1β作为NLRP3炎症小体的下游信号分子,参与AMD的发生,其可能成为AMD未来的治疗靶点。
IL-18在AMD发展过程中的作用较复杂,不同研究得出的结论并不相同。DICER1缺陷引起Alu RNA积累可激活NLRP3炎症小体,导致人RPE细胞中IL-18分泌增加,中和IL-18可以逆转RPE细胞变性,提示IL-18是Alu RNA介导RPE细胞毒性的效应分子[28]。然而,有研究显示IL18-/-小鼠(类似于NLRP3-/-)在激光诱导条件下产生更严重的CNV,在小鼠玻璃体内或全身注射IL-18可以减轻激光诱导的CNV,同时对RPE细胞活性及其屏障完整性无明显影响[46-47]。最近有研究发现,IL-18并不会影响CNV的发展,仅仅会引发干性AMD改变[48]。因此,在靶向炎症小体进入临床试验之前,严格评估IL-18或NLRP3炎症小体是否影响RPE细胞活性和CNV发展至关重要。
4 以NLRP3炎症小体为靶向的潜在治疗策略
4.1 抑制P2X7和NF-κB
研究表明P2X7或NF-κB低表达小鼠经Alu RNA诱导后RPE损伤较轻,提示NF-κB及P2X7嘌呤受体可能成为AMD的治疗靶点[33]。核苷类逆转录酶抑制剂和丹参酚酸A被发现可以抑制P2X7介导的RPE损伤和GA[49-50]。Liu等[51]研究发现,长春西汀可以通过抑制NF-κB途径来抑制TNF-α、IL-1β、IL-18等炎性因子的产生,可能作为AMD的潜在治疗药物。
4.2 抑制MyD88
在Alu RNA诱导的RPE损伤过程中,MyD88同时起到激活NLRP3炎症小体和参与IL-18下游信号转导的作用,通过IL-1受体相关激酶1/4介导RPE细胞损伤,抑制MyD88可以减轻Alu RNA诱导的视网膜损伤[28]。由此可见,MyD88靶向治疗既可以抑制NLRP3炎症小体生成,又可以影响IL-18的作用。
4.3 抑制IL-18
目前,IL-18对AMD的作用仍存在争议。Tarallo等[28]认为IL-18的表达增加可能加重早期AMD的发展和GA,但Doyle等[46]研究发现,注射纯化的IL-18与抗VEGF治疗联合使用时对新生血管性AMD治疗作用更加明显。虽然不同研究得出的结论不同,但IL-18仍有可能成为AMD的潜在治疗靶点。未来需要更多的研究来进一步明确IL-18在AMD,尤其是CNV发展中的作用。
4.4 其他治疗策略
有研究将NLRP3炎症小体激活通路上其他组分作为AMD治疗的靶点,包括用PTX3结合补体因子H、格尔德霉素和齐多夫定阻止NLRP3炎症小体激活,花青素3-葡萄糖苷抑制4-羟基己烯醛对NLRP3炎症小体的激活,人肝脏X受体抑制β-淀粉样蛋白对NLRP3炎症小体的激活等[52-56]。
5 小结与展望
NLRP3炎症小体与AMD发生的相关性逐渐得到验证,但其具体作用机制仍未十分明确。正是由于NLRP3炎症小体及其下游因子作用机制复杂使得针对其的靶向治疗及药物研发趋于困难。目前临床上湿性AMD的治疗主要为抗VEGF药物,但其价格昂贵,而对于干性AMD临床上至今尚无有效的应对措施。因此,需要继续致力于NLRP3炎症小体的研究,未来需更深入探讨NLRP3炎症小体信号通路在AMD发生和发展中的主要作用靶点。随着其作用机制的逐步揭开,针对NLRP3炎症小体的治疗将在未来发挥越来越重要的作用。NLRP3炎症小体有望为AMD特别是干性AMD的防治提供新的关键作用靶点和治疗措施。
利益冲突所有作者均声明不存在利益冲突
