同一患儿罹患3种遗传性肝病报道
2022-02-12纪培林宋元宗
纪培林,宋元宗
(暨南大学附属第一医院 儿科,广东 广州 510630)
对于迁延性黄疸伴肝大,尤其是直接胆红素、总胆汁酸和γ-谷氨酰转肽酶升高的患儿,应警惕遗传性肝病并积极进行遗传学分析明确病因。遗传性肝病经积极对症治疗,也有预后良好的可能。本文旨在报道1例同时罹患钠牛磺胆酸共转运多肽(Na+-taurocholate cotransporting polypeptide,NTCP)缺陷病、Alagille综合征和Gilbert综合征3种遗传性肝病患儿的临床和分子遗传学特点,以提高临床医生对此类疾病的认识。
1 病例资料
患儿,男,2月龄,因“皮肤巩膜黄染近2个月”至暨南大学附属第一医院门诊就诊。家长诉患儿出生后第3天即开始出现皮肤、巩膜黄染。出生后40 d时因“皮肤黄染38 d”在当地医院住院治疗,期间发现总胆红素升高,以直接胆红素为主,总胆汁酸、γ-谷氨酰转肽酶等指标也升高(具体数据见表1,1.5月龄时)。红细胞葡萄糖-6-磷酸脱氢酶活性、乙肝两对半(定性)、肝炎病毒3项(甲肝病毒、丙肝病毒和戊肝病毒IgM抗体)、致畸4项、甲功5项、EB病毒及巨细胞病毒DNA检测均未见异常。肝胆胰脾彩超提示肝内外胆管未见扩张,肝脾未见明显占位。诊断为婴儿胆汁淤积症,采用葡醛内酯、熊去氧胆酸及丁二磺酸等药物治疗。治疗3 d后,皮肤黄染未见明显消退而自行出院。2月龄时因黄疸未消退首次来暨南大学附属第一医院门诊就诊。发病以来,未解陶土样大便,小便正常。
体格检查:头围38 cm,身长59.4 cm,体质量4.93 kg;双眼巩膜、颜面及躯干部皮肤黄染;头颅五官无畸形;双肺呼吸音清,两侧对称,未闻及干湿性啰音;心音有力,各瓣膜区未闻及病理性杂音;腹平软,肝肋下3 cm,质软,脾肋下未触及;四肢无畸形,肌张力正常。
患儿系第4胎第4产,孕36+6周顺产出生,出生体质量3.45 kg,身长50 cm。其父母均体健,均否认乙肝病史,但其父自诉有皮肤巩膜黄染史多年,但生化检查未见异常(见表1)。
患儿查血生化发现总胆红素升高,以直接胆红素为主,伴总胆汁酸、γ-谷氨酰转肽酶升高,锌离子、25-羟基维生素D及铁蛋白水平均降低,而甲胎蛋白明显升高(见表1)。心脏超声和脊柱全长正侧位片均未见异常,眼角膜后胚环检查阴性。
根据临床表现和检查诊断为胆汁淤积症、维生素D缺乏以及锌缺乏症。给予患儿口服苯巴比妥、补充骨化三醇、维生素AD及锌等对症支持治疗。
为评估遗传性胆汁淤积症的可能,经医院医学伦理委员会批准及监护人签署知情同意书后对患儿及其父母进行高通量测序。经Sanger测序验证,患儿表现出以下3种基因变异(见图1):(1)JAG1基因变异c.1511A>G(p.Asn504Ser)杂合子,变异来自其父;(2)SLC10A1基因变异c.800C>T(p.Ser267Phe)纯合子,变异来自其父母;(3)UGT1A1基因变异c.211G>A (p.Gly71Arg)纯合子,变异来自其父母。其父为同样基因型,而其母为SLC10A1基因和UGT1A1基因变异携带者(见图1)。以上变异均为文献[1-4]报道的致病性变异。因此,确诊为NTCP缺陷病、Alagille综合征及Gilbert综合征这3种遗传性肝病。
确诊遗传性肝病后,继续给予患儿口服苯巴比妥、骨化三醇、维生素AD及补锌等对症支持治疗。门诊规律随访至9月龄,患儿黄疸逐渐消失,除总胆汁酸外,其余异常生化指标逐渐改善(见表1),且头围、体质量和身长等体格发育指标均正常。
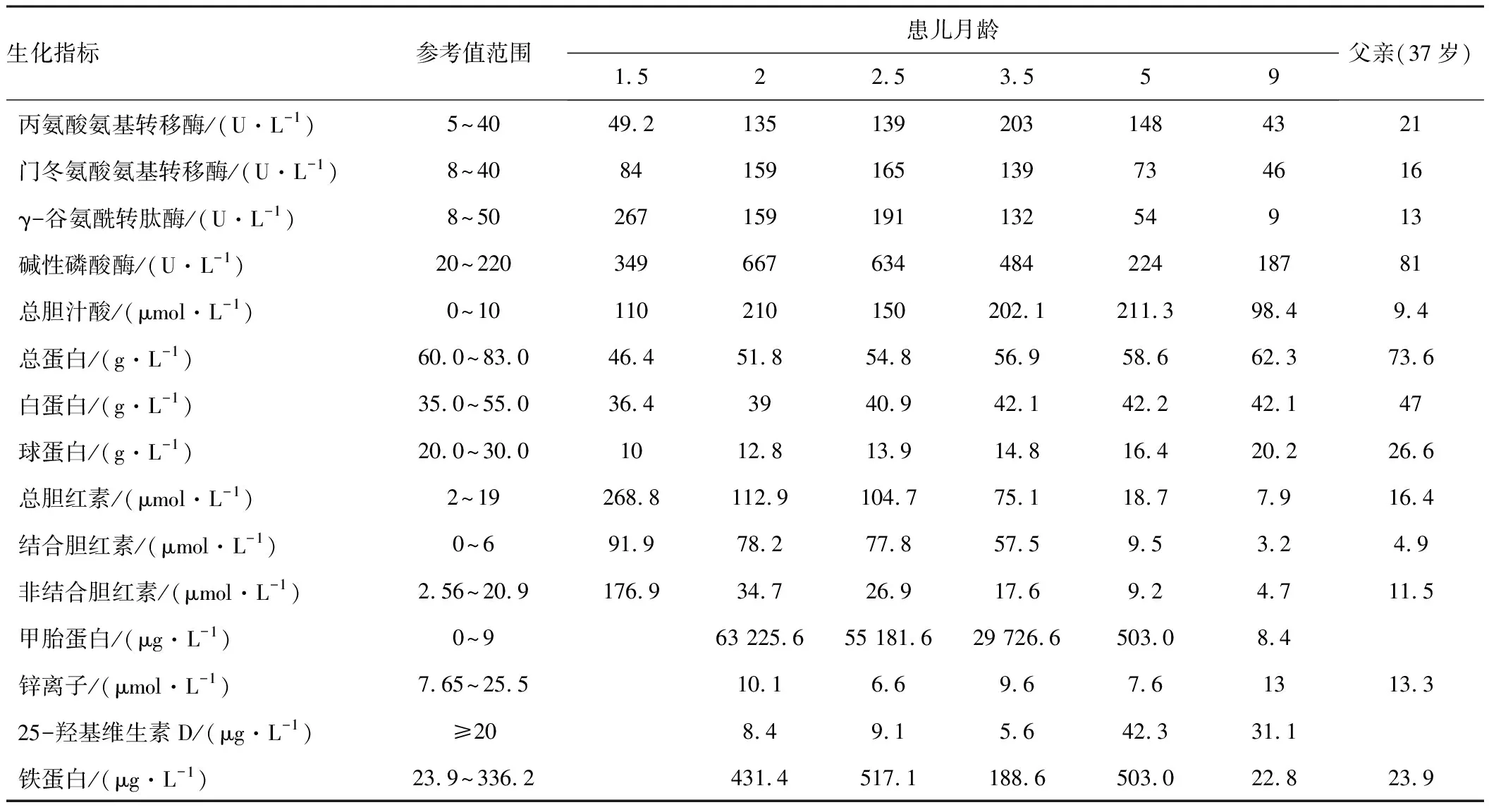
表1 患儿及其父生化指标动态变化情况
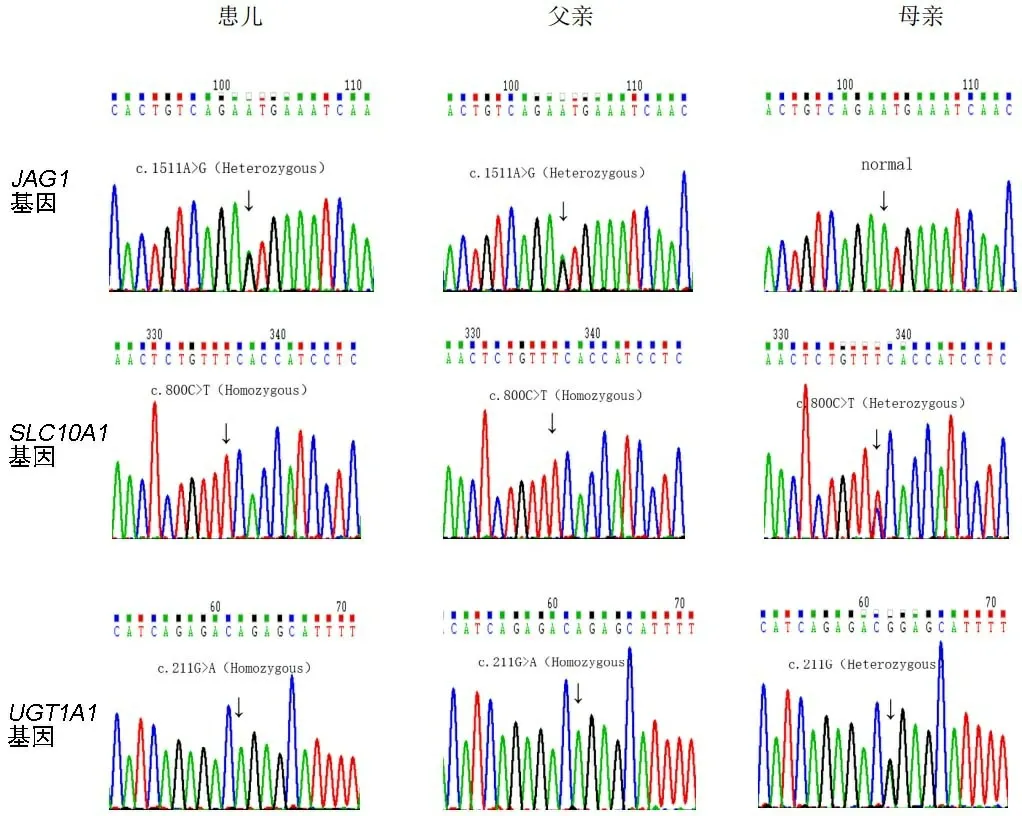
杂合为Heterozygous,纯合为Homozygous,正常为normal。
2 讨论
本研究患儿以黄疸为突出表现,病程迁延,总胆红素升高,以直接胆红素水平升高为主,伴γ-谷氨酰转肽酶明显升高,胆汁淤积症诊断成立。胆汁淤积症的病因复杂,随着基因组学和分子遗传学理论及技术的发展、成熟和临床应用,近年来遗传性胆汁淤积症的诊断治疗引起了儿科界的关注[5]。经过遗传学分析,结合上述临床表现,本研究患儿被确诊NTCP缺陷病、Alagille综合征和Gilbert综合征这3种遗传性肝病,从而明确了胆汁淤积症的病因,并为治疗方案的制定和预后评估提供了遗传学依据。
NTCP缺陷病是位于染色体14q24.2的SLC10A1基因突变引起的遗传性胆汁酸代谢病[6-7],患者突出临床特点为高胆汁酸血症,但部分患儿表现为新生儿期高胆红素血症和婴儿早期一过性胆汁淤积症[8-9]。NTCP是一种表达于肝细胞基侧膜的钠依赖性转运蛋白,其主要功能是作为溶质载体10家族的一部分,以钠依赖方式将结合型胆汁酸从血浆摄入肝细胞中,在胆汁酸的肠肝循环中起重要作用[10-13]。人类SLC10A1基因c.800C>T(p.Ser267Phe)变异将导致NTCP对胆汁酸摄取功能几乎完全丧失[14]。本研究患儿系SLC10A1基因变异c.800C>T的纯合子,NTCP诊断成立。部分牛磺胆酸共转运多肽缺陷病患儿存在血清锌离子及25-羟基维生素D缺乏,此类患儿25-羟基维生素D水平降低可能与肠道内胆汁酸减少和(或)活性不足导致膳食脂肪溶解和脂溶性维生素吸收减少有关[15]。本例患儿经过补充骨化三醇、维生素AD及锌等处理后,血清中锌离子、25-羟基维生素D及铁蛋白逐渐恢复至正常水平。
Alagille综合征是以慢性胆汁淤积为突出临床特征的常染色体显性遗传性疾病,常累及多个系统[16]。近95%的Alagille综合征为位于染色体20p12的JAG1基因突变引起[2]。该病经典的诊断标准需同时满足慢性胆汁淤积、心脏杂音、蝴蝶椎骨、角膜后胚胎环和特殊面容五大临床表现[17],但并非所有的Alagille综合征患者都同时满足上述5条典型诊断标准[18]。Kamath等[19]和Guru等[20]提出了修订的Alagille综合征诊断标准,若患儿存在JAG1基因突变同时有家族阳性病史,即使无胆管稀疏及心脏、肾脏、眼部、脊柱、面部的临床表现也可诊断。本研究患儿仅有慢性胆汁淤积,无其他四大临床表现,但遗传学分析发现JAG1基因致病性突变c.1511A>G(p.Asn504Ser)。根据Alagille综合征的修订诊断标准,患儿可确诊为Alagille综合征。
Gilbert综合征是UGT1A1基因突变影响肝细胞内尿苷二磷酸-葡萄糖醛酸转移酶活性,致使非结合胆红素葡萄糖醛酸化障碍而形成的常染色体隐性遗传性病[21-22]。该病是一种良性的、可自行消退的黄疸,除非结合胆红素升高外,其他肝功能指标正常且不存在溶血[22]。本研究患儿存在UGT1A1基因c.211G>A(p.Gly71Arg)纯合变异,遗传方式符合常染色体隐性遗传,可确诊Gilbert综合征。文献报道该病并发其他疾病可加剧黄疸程度[23]。本研究患儿同时患有NTCP缺陷病及Alagille综合征,可能进一步延长黄疸消退时间,但最终患儿非结合胆红素逐渐降至正常范围。
Alagille综合征患儿由于肝内胆管稀疏,以肝内胆汁淤积症为突出临床表现,胆红素升高以直接胆红素为主[24]。NTCP缺陷病则与新生儿期高间接胆红素血症和婴儿早期胆汁淤积症有关[9]。Gilbert综合征以间接胆红素升高为主[23]。本研究患儿同时罹患3种遗传性肝病,三者相互协同,最终形成以黄疸为主的临床表现,且病情迁延,总胆红素升高,以直接胆红素升高为主,同时伴总胆汁酸、γ-谷氨酰转肽酶明显升高。
本研究患儿虽然患有3种遗传性肝病,但经规律门诊复查和对症支持处理,最终生化指标恢复正常,体格发育良好。其父具有同样基因型,但就诊时缺乏明显临床表现,化验结果未见异常。该病例给临床医生的启示是,在临床工作中对于迁延性黄疸,尤其直接胆红素升高为主的患儿,应警惕遗传性肝病的可能,并积极进行遗传学分析查找病因。遗传性肝病未必是不治之症,甚至是罹患多种遗传性肝病的患儿也有预后良好的可能性,医务人员和家长对此要有充分信心。
