基因A型和B型牛鼻病毒双重荧光定量RT-PCR检测方法的建立及应用
2022-02-04周昱行
周昱行,汤 承,岳 华
(西南民族大学畜牧兽医学院,四川 成都 610041)
牛鼻病毒(Bovine rhinitis virus,BRV)是小RNA病毒科口蹄疫病毒属成员,为无囊膜单股正链RNA病毒[1]。该病毒引起牛鼻甲、气管和肺上皮的损伤,是牛呼吸道疾病综合征(Bovine respiratory dis⁃ease complex,BRDC)的重要病原[2-4]。根据病毒基因组序列,可将BRV 划分为基因A 型(BRAV)和基因B型(BRBV)2 个基因型[5],BRAV 的致病性比BRBV强[6]。对犊牛人工感染BRAV 后,感染牛出现了发热、精神沉郁、局灶性鼻炎、肺炎等典型的呼吸道症状[6-8]。如今,BRV 已经广泛分布于欧洲、亚洲、美洲和非洲共9 个国家,检出率最高可达52.7%[9],成为牛呼吸道疾病的重要病原[10]。
BRV 基因组全长约7.2 kb~7.5 kb,编码4 个结构蛋白(VP1~VP4)和8 个非结构蛋白(Lpro、2A、2B、2C、3A、3B、3C 和3D),其3D 基因相对保守,是目前BRV 最常用的检测靶基因,但其在2个基因型间差异较大,二者基因序列平均同源性仅为55.8%,无法设计检测BRV的通用型PCR引物。目前已发表的4种BRV的检测方法均为靶向检测BRAV或BRBV 3D基因的单一荧光定量RT-PCR[10-13],尚无同时检测BRAV和BRBV 的双重荧光定量RT-PCR 方法。由于小RNA 病毒5'非翻译区(5'NTR)含内部核糖体进入位点(IRES),介导病毒基因组与宿主细胞内核糖体的结合[14-15],3'非翻译区(3'NTR)包含与负链RNA 合成有关的顺式作用元件,这2 个区域的基因序列分别在BRAV 和BRBV 中较为保守[16-17]。本研究对GenBank登录的全部9 条BRAV 基因序列和全部21 条BRBV 基因序列进行了比对分析,发现BRAV 5'NTR 序列的平均同源性为91.7%,其3D 基因序列的平均同源性为83.8%;而BRBV 3'NTR 序列的平均同源性为95.67%,其3D 基因序列的平均同源性为86.48%,表明NTR 更适合作为BRV 的分子检测靶标。本研究根据BRAV 5'NTR 和BRBV 3'NTR 分别设计引物和探针,建立同时检测BRAV 和BRBV 的双重荧光定量RT-PCR 方法,并将其应用于临床样品的检测,为BRDC 的诊断和防控提供了参考。
1 材料与方法
1.1 病毒/细菌核酸和临床样品BRAV 和BRBV 核酸由本实验室检测并经测序鉴定;D 型流感病毒(IDV)、牛冠状病毒(BCoV)、牛病毒性腹泻病毒1 型(BVDV-1)、牛呼吸道合胞体病毒(BRSV)、牛副流感病毒3 型(BPIV-3)、牛腺病毒3 型(BAdV-3)、牛支原体、多杀性巴氏杆菌、溶血曼氏杆菌、大肠杆菌和沙门菌等病原的核酸样品由西南民族大学动物医学实验室保存。43 份BRDC 病牛鼻拭子样品于2020 年10 月~2021 年10 月采自四川省、河北省和吉林省,其中32 份来自四川省的2 个牛场,8 份来自河北省的1 个牛场,3 份来自吉林省的1 个牛场,所有样品于-80 ℃保存。
1.2 载体及主要试剂pMD19-T载体、QIAamp Viral RNA Mini Kit 购 自QIAGEN 公 司;Premix ExTaq购 自TaKaRa 公司;胶回收试剂盒、Maxima H Minus Dou⁃ble-Stranded cDNA Synthesis Kit 购自ThermoFisher Sci⁃entific 公司;QuickTaqHS DyeMix 购自TOYOBO 生物科技有限公司;大肠杆菌DH5α 感受态细胞购自北京擎科生物科技有限公司。
1.3 引物及探针的设计、合成与特异性检测对GenBank 中9 条BRAV 基 因 序 列 和21 条BRBV 基 因序列经MEGA7 程序比对分析后,针对BRAV 5'NTR 保守区和BRBV 3'NTR保守区分别设计特异性引物及不同荧光基团标记的探针(表1)。BRAV 的预期扩增片段为144 bp,位 于402 bp~546 bp(KT948520);BRBV的预期扩增片段为92 bp,位于7 429 bp~7 521 bp(EU236594)。引物由北京擎科生物科技有限公司合成。
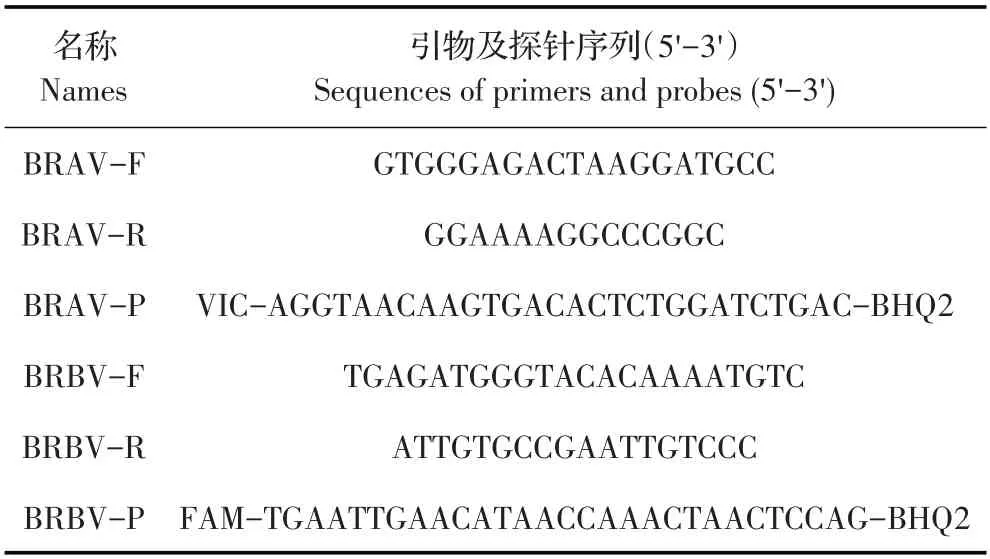
表1 荧光定量PCR方法的引物和探针信息Table 1 Primers and probes for real-time quantitative RT-PCR assays
1.4 重组质粒标准品的构建与鉴定以表1 中的BRAV-F/R、BRBV-F/R 引物分别对BRAV、BRBV 的核酸样品进行PCR 扩增,将PCR 产物分别克隆至pMD19-T 载体,构建重组质粒pBRAV 和pBRBV,经PCR 鉴定后由京擎科生物科技有限公司测序鉴定。使用NanoDrop微量分光光度计测定重组质粒浓度,参照Wu等的方法计算重组质粒标准品的拷贝数[18]。
1.5 双重荧光定量RT-PCR 反应条件的优化及标准曲线的建立采用20 μL 体系:重组质粒标准品pBRAV 和pBRBV 各0.5 μL,Premix ExTaq10 μL,BRAV-F/R、BRBV-F/R(10 μmol/μL)各1.0 μL,2 种探针(10 μmol/μL)各0.1 μL,ddH2O 补足至20 μL。分别以47 ℃、48 ℃、49 ℃、50.2 ℃、51.2 ℃、52 ℃退火,以二者扩增的Ct 值最小时的温度为最佳退火温度。在最佳退火温度下,将BRAV 和BRBV 引物浓度设置为0.125 mol/L~1.000 mol/L,探针浓度设置为0.025 mol/L~0.150 mol/L,通过棋盘法优化两种探针和引物的浓度。
以稀释度为101~105的重组质粒标准品各0.5 μL作为模板,设置3 组平行对照,利用建立的双重荧光定量RT-PCR 方法检测,取3 个重复组的平均值,使用R 语言计算Ct 值与拷贝数的拟合优度,并使用ggplot2 绘图包绘制标准曲线,通过公式E=10(-1/k)-1 计算扩增效率。
1.6 特异性试验以本实验室保存的BRAV、BRBV、IDV、BCoV、BVDV-1、BRSV、BPIV-3 的cDNA 以及BAdV-3、牛支原体、多杀性巴氏杆菌、溶血曼氏杆菌、大肠杆菌和沙门菌的DNA 为模板,以pBRAV+pBRBV 重组质粒标准品作为阳性对照,以ddH2O 作为阴性对照,利用建立的双重荧光定量RTPCR 方法扩增,评估该方法的特异性。
1.7 敏感性试验以稀释度为100~109的重组质粒标准品pBRAV、pBRBV 各0.5 μL 作为模板,利用建立的双重荧光定量RT-PCR 方法扩增,并设立ddH2O 为阴性对照,当Ct 值≤40 时判定为阳性结果,评估该方法的敏感性。
1.8 重复性试验批内重复性试验:分别取稀释度为100~105的重组质粒标准品pBRAV、pBRBV各0.5 μL作为模板,利用建立的双重荧光定量RT-PCR 扩增,每个浓度设置3 个重复;批间重复试验:在不同的时间相同的反应条件下对上述稀释度的重组质粒标准品经该荧光定量RT-PCR 扩增,每种浓度设置3 个重复。通过计算批内、批间变异系数评估该方法的重复性。
1.9 临床样品的检测将43份BRDC病牛鼻拭子样品每份加入5 mL PBS溶液混匀,涡旋3 min,10 000 r/min离心3 min 后取上清;以QIAamp Viral RNA Mini Kit提取病毒RNA,反转录为cDNA 后作为模板,以本研究建立的双重荧光定量RT-PCR 方法检测。由于目前尚无OIE 指定的BRV 检测方法,因此本研究采用已报道的3 种BRAV 检测方法[10,12-13]和4 种BRBV 检测方法[10-11,13]对上述临床样品进行检测,并比较本研究所建方法与各方法的检测结果。
2 结 果
2.1 重组质粒标准品的鉴定结果构建的重组质粒标准品pBRAV 和pBRBV 经PCR 鉴定结果显示,分别经PCR扩增出长度为144 bp和92 bp的目的片段(图1),均与预期符合。测序结果显示,插入的144 bp 片段与BRAV_RS3X 株(KT948520)5'NTR 基因序列的同源性为99.3%,插入的92 bp 片段与BRBV_BSRI3(KP264975)3'NTR 基因序列的同源性为100%,表明正确构建了重组质粒标准品pBRAV 和pBRBV,浓度分别为27.00 ng/μL 和79.67 ng/μL。将两种重组质粒标准品均稀释至1.0 ng/μL,经计算,二者的拷贝数均为3.2×109拷贝/μL。

图1 重组质粒标准品的PCR鉴定结果Fig.1 Identification of recombinant plasmid standard by PCR
2.2 反应条件的优化及标准曲线的绘制通过棋盘法优化双重荧光定量RT-PCR的反应体系,20 μL反应体系为:Premix ExTaq10 μL,BRAV-F/R(10 μmol/μL)各1 μL,BRBV-F/R(10 μmol/μL)各3 μL,BRAV-p、BRBV-p(均10 μmol/μL)各0.25 μL,质粒标准品各0.5 μL,补足ddH2O 至20 μL;优化后的反应程序为:95 ℃2 min;95 ℃15 s、50.2 ℃20 s,共40个循环。
线性回归分析结果显示,pBRAV 和pBRBV 扩增的Ct 值与其拷贝数的拟合优度均达到0.99,线性关系良好。线性方程分别为:BRAV:y=40.3-3.0x,R2=0.99;BRBV:y=42.5-3.2x,R2=0.99。扩增效率为:EBRAV=115.4%,EBRBV=105.4%(图2)。结果表明两种重组质粒标准品在101~105稀释度即3.2×108拷贝/μL~3.2×104拷贝/μL浓度下均与各自的Ct 值呈良好的线性关系。
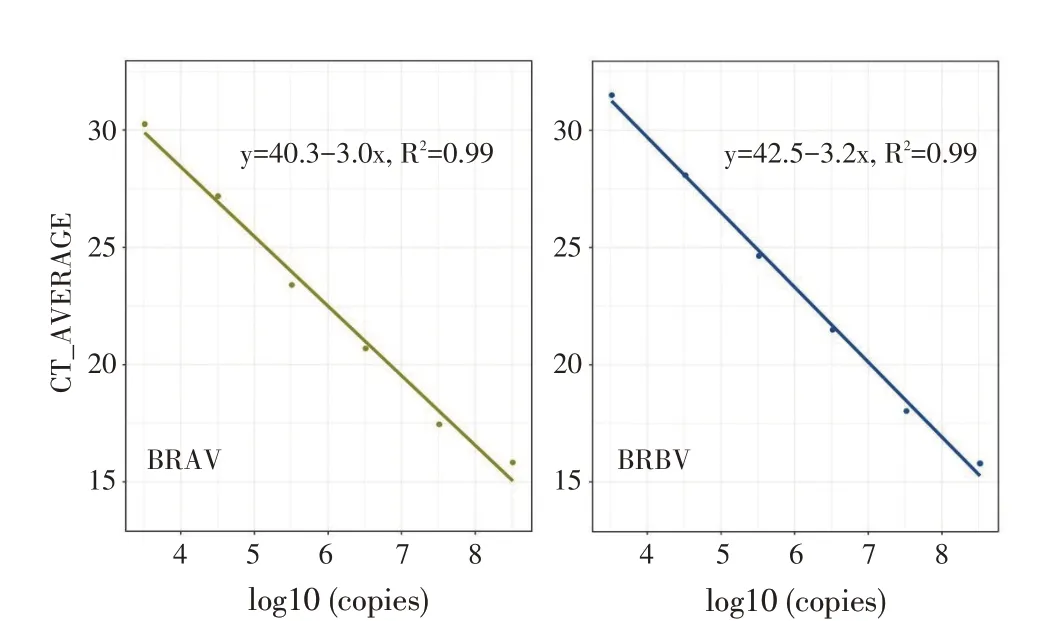
图2 双重荧光定量RT-PCR 检测方法的标准曲线Fig.2 Standard curves of duplex quantitative real-time RT-PCR
2.3 特异性试验结果特异性试验结果显示,本研究建立的双重荧光定量RT-PCR 方法仅特异性扩增重组质粒标准品pBRAV 和pBRBV,对IDV、BCoV、BVDV-1、BRSV、BPIV-3、BAdV-3、牛支原体、多杀性巴氏杆菌、牛溶血曼氏杆菌、大肠杆菌和沙门菌均无扩增曲线(图3),表明本研究建立的双重荧光定量RT-PCR 方法特异性较强。

图3 双重荧光定量RT-PCR的特异性试验结果Fig.3 Specificity test of duplex quantitative real-time RT-PCR assay
2.4 敏感性试验结果以建立的双重荧光定量RTPCR 方法对100~109(3.2×109拷贝/μL~3.2×100拷贝/μL)稀释度重组质粒标准品进行扩增,确定该方法的敏感性。结果显示,该方法对BRAV、BRBV 质粒标准品的检测下限均为3.2×101拷贝/μL(图4),表明本研究建立的双重荧光定量RT-PCR 方法敏感性较高。

图4 BRAV(A)、BRBV(B)双重荧光定量RT-PCR的敏感性试验结果Fig.4 The sensitivity test of the duplex quantitative real-time RT-PCR for BRAV(A)and BRBV(B)
2.5 重复性试验结果取稀释度分别为100~105即3.2×109拷贝/μL~3.2×104拷贝/μL 的重组质粒标准品pBRAV、pBRBV 进行批内和批间重复性试验。结果显示:本研究建立的双重荧光定量RT-PCR 检测方法批内和批间重复性试验Ct 值的变异系数分别为0.33%~2.46%及1.42%~3.92%,均小于5%(表2),表明该双重荧光定量RT-PCR 方法的重复性较好。

表2 双重荧光定量RT-PCR方法的重复性试验结果(n=3)Table 2 Repeatability evaluation of duplex quantitative real-time RT-PCR(n=3)
2.6 临床样品检测结果利用国外文献报道的3 种BRAV 检测方法和4 种BRBV 检测方法以及本研究所建方法同时检测43 份BRDC 病牛鼻拭子样品。结果显示,本研究建立的方法对BRAV 和BRBV 的检出率分别为32.56%(14/43)和30.23%(13/43),二者混合感染率为22.73%(5/22),首次证实了国内牛群存在BRAV 的感染;其他3 种BRAV 检测方法的检出率分别为0(0/43)、2.33%(1/43)、23.26%(10/43),其他4种BRBV检测方法的检出率分别为27.91%(12/43)、27.91%(12/43)、27.91%(12/43)、27.91%(12/43)。本研究所建方法对BRAV 和BRBV 的检出率均高于所有已有的检测方法,表明该方法可以用于临床BRV的检测。
3 讨 论
BRV 是引起BRDC 的重要病原[12,19],由于具有2种基因型,并且2型混合感染较为普遍,如Ng等报道的美国BRDC 患牛中2 型BRV 的混合感染率为7%[10],而现有的方法均是基于单一PCR 对2 种基因型BRV 分别检测[10-13]。本研究分析BRV 基因序列时发现其NTR 最为保守,更适合作为检测靶点,因此本研究针对BRAV 5'NTR 和BRBV 3'NTR 分别设计引物和探针,并经各反应条件的优化建立了同时检测BRAV和BRBV 的双重荧光定量RT-PCR 方法,且经试验证明该方法特异性强、敏感性高、重复性好。
为进一步了解BRAV的分子特征并分析已报道的方法对BRDC 病牛鼻拭子样品BRAV 检出率低的原因,本研究从检出的BRAV 阳性样品中经PCR 扩增了3 个近乎全长的BRAV 基因组(MZ004694~MZ004696)(未发表),比对分析结果显示3 种方法报道的引物均存在与扩增序列不完全匹配的现象,如Ng 等所建方法下游引物3'端的4 个碱基中有3 个碱基与参考序列的对应位点不匹配,且第4 个碱基与3 株国内BRAV相应位点碱基均不匹配[10];Kishimoto 等报道的BRAV下游引物3'端第3 个碱基与3 株国内BRAV 参考序列对应位点的碱基均不匹配[13],这些可能是导致这两种方法对BRAV 检出率低的原因。Hause 等所建方法下游引物3'端的6 个碱基中存在3 个碱基与参考序列对应位点碱基不匹配[12],且第2 个、第6 个碱基与上述3 株国内BRAV 相应位点碱基均不匹配,可能是导致该方法未有效检出国内BRAV 的原因。这些结果表明上述3 种方法均不适用于国内BRAV 的检测。而本研究设计的BRAV 引物与3 株国内BRAV 相应基因序列完全匹配,且BRBV 引物3'端17 个碱基与所有BRBV 参考株相应基因序列均匹配,因此建立的双重荧光定量RT-PCR 方法扩增效率及敏感性均较高。该方法使用不同荧光基团修饰的探针,可以同时检测两型BRV,特异性较强,并且可在1 h 左右完成检测,与恒温热隔绝式RT-PCR 用时相近[20],为BRV 提供了快捷的检测手段。上述结果提示,进一步加强国内BRV 流行株的分子特征研究将有助于BRV 检测方法的优化和完善。
近年来,国内肉牛养殖业发展迅速,呼吸道疾病对养牛业危害大,已成为影响肉牛养殖业经济效益的重要原因[21]。然而国内对BRDC 的研究较晚,且其流行病学资料较少,虽已证实牛传染性鼻气管炎病毒(IBRV)、BPIV-3、BCoV、BVDV 等是BRDC 的重要病原[22],但对致BRDC 的BRV 还知之甚少。最近Xie 等报道了BRBV 在国内的存在[11],但目前国内尚无BRAV 的相关报道。本研究利用建立的双重荧光定量RT-PCR 方法检测四川、河北和吉林省4 个不同地区的43份BRDC病牛鼻拭子样品,并与已报道的3 种BRAV 及4 种BRBV 的荧光定量RT-PCR 方法进行了比较。结果显示,本研究建立方法对BRV的检出率为51.16%(22/43),其中BRAV 和BRBV 的检出率分别为32.56%(14/43)和30.23%(13/43),均高于所有已报道的BRV检测方法。也表明BRV是BRDC的重要病原,并首次证实BRAV 在国内的存在,丰富了BRV 的分子流行病学资料。值得注意的是,在22份BRV阳性样品中,5份样品存在BRAV和BRBV的混合感染,感染率为22.73%(5/22),该现象与Ng 等在加利福尼亚州报道的结果相近[10],进一步表明建立检测BRAV 和BRBV 双重荧光定量RT-PCR 方法的必要性。
综上所述,本研究建立了同时检测BRAV 和BRBV 的双重荧光定量RT-PCR 方法,该方法具有特异性强、敏感性高、稳定性和重复性好的特点,为BRV 的检测提供了新的检测手段,并首次证实国内牛群中存在BRAV 的感染,且还有BRAV 和BRBV 混合感染的现象,为国内BRDC 的防控提供了新的参考依据。
