氮化硅粉体制备技术及粉体质量研究进展
2022-01-26向茂乔耿玉琦朱庆山
向茂乔,耿玉琦,2,朱庆山,2
(1中国科学院过程工程研究所多相复杂系统国家重点实验室,北京 100190;2中国科学院大学化学工程学院,北京 100049)
引 言
氮化硅(Si3N4)是典型强共价键化合物,不仅熔点高、硬度大、耐磨损,而且抗弯强度高、导热性能好[1],在国防、军工、电子信息等关键领域具有不可替代的地位[2],关乎国家安全和国民经济的发展。例如,Si3N4陶瓷可作为超声速导弹的天线罩[3-4],战斗机和航空发动机的精密轴承球及轴承环[5-6],封装芯片的基板[7-8]等。其中,仅Si3N4轴承球2019年全球的消费总额就达到4.7 亿美元,年增长率约保持在8.0%。但现阶段我国高性能Si3N4轴承球对外依存度超过了90%,国际市场几乎被美国CoorsTek 和日本东芝陶瓷所垄断[5]。此外,在集成电路领域,集成度和功率越来越大,对封装芯片的基板抗弯强度、稳定性、散热能力都提出了更高的要求[9]。Si3N4基板比传统Al2O3和AlN 陶瓷基板具有更高的抗弯强度(600~800 MPa)、更优异的防潮能力和更好的循环稳定性(≥5000次)。但由于Si3N4基板的造价较高,目前只在轨道交通、风电、光伏、新能源汽车等的IGBT(insulated gate bipolar transistor)功率模块得到小规模应用[7-9]。日本东芝(Toshiba)、日本京瓷(Kyocera)、德国Curamik 公司、美国罗杰斯(Rogers)率先制备出高性能Si3N4基板[热导率约110 W/(m•K),抗弯强度约650 MPa],并使IGBT 功率模块最高服役温度从125℃提升至200℃,相比于AlN基板,Si3N4基板寿命延长了10倍之多,且在-40~150℃之间热循环次数提升了25 倍,在全球具有压制性的优势。日本东芝2020 年投资建设Si3N4基板生产基地,预计2023 年3月建设完成,产能可达40000 m2/a。我国Si3N4基板目前仍处于初始研发阶段。根据中国产业研究院统计,全球Si3N4基板的需求量在未来五年内将以3.29%的平均增长率增长,在2022年将增加到30200 m2,仅我国IGBT模块市场规模将达到200亿元。因此可以预计,在不久的未来Si3N4将迎来高速发展。
Si3N4粉体是制备陶瓷的基础。根据不同用途可把氮化硅粉体分为三大类:陶瓷级粉体,光伏级粉体,电子级粉体。陶瓷级粉体主要用于制备结构陶瓷,例如防弹片、轧辊、轴承球、刀具、升液管等;光伏级粉体主要用作多晶硅铸锭工艺中的脱模剂(喷涂在坩埚内壁);电子级粉体主要用于制备陶瓷基板。高质量粉体是制备高性能Si3N4陶瓷的首要前提[8,10]。在不同的领域中,对粉体质量的要求也不一致。对于多晶硅领域的脱模剂来说,光伏级粉体需要满足纯度高和流动性好的要求。对于制备抗弯强度大且热导率高的陶瓷基板和高性能轴承球来说,陶瓷级粉体和电子级粉体不仅需要纯度高,而且还需要满足低氧、超细、高α相等指标。因为这些指标都会直接决定基板和陶瓷球中的缺陷(晶格氧、气孔)、杂质以及晶界尺寸,从而影响热导率和抗弯强度。最近,日本宇部(UBE)探究了粉体质量对陶瓷热导率和抗弯强度的影响行为[11-12],发现当粉体粒径D50为0.4~1.5 μm,比表面积(BET)为4.0~9.0 m2/g,α 相含量>95%(质量),O含量为0.2%~0.95%(质量),C含量<0.2%(质量),F含量<0.003%(质量),Cl含量<0.01%(质量),以及Ca+Fe+Al等其他金属总杂质<0.05%(质量)时,具有较高的烧结活性,可制备出热导率>100 W/(m•K),抗弯强度大(>1000 MPa)的高性能基板[商业化应用的最低热导率和抗弯强度为90 W/(m•K)和600 MPa]。通常,粉体质量与其制备方法密切相关。本文从反应原理综述了四种不同的制备方法(SiO2碳热氮化法,Si 粉直接氮化法,化学气相合成法,硅胺前体高温热分解法),着重总结了这四种方法在优化传质与传热改善粉体质量方面的现状,并介绍了国内外主要企业生产粉体质量的现状,展望了连续批量化制备高质量粉体的发展趋势和方向。
1 Si3N4粉体制备技术及粉体质量的研究现状
1.1 SiO2碳热氮化法
SiO2碳热氮化法制备Si3N4粉体的反应原理是在1400℃以上激活C 粉的还原性,将SiO2粉体还原并氮化为Si3N4,如式(1)所示。然而,SiO2-C-N2体系的反应并不是单一反应,而是多个反应共存的复杂反应,除SiO2、C、N2原料参与反应外,中间产物SiO 和CO 也会参与反应,不仅可以生成Si3N4,也可以生成SiC和Si2N2O,部分副反应如反应式(2)~式(8)所示。

根据热力学分析可知,SiO2-C-N2体系产物的组成与温度、N2压力、CO 分压密切相关,如图1 所示,在特定的温度和CO 分压下可获得单相Si3N4(s)。但是,从动力学角度考虑,万小涵等[13]发现,生成SiC和Si3N4边界温度差异小,SiC 杂相不可避免。此外,整个合成反应过程中,同时存在固-固反应、气-固反应、气-气反应。对于固相参与的反应,初始生成的Si3N4、SiC 或Si-O-N 固溶体会覆盖在原料表面形成传质障碍层[14-18],其扩散传质的活化能较大,约306 kJ/mol,导致后续氮化难度大[18],产物中经常会存在未反应完全的Si2N2O中间物或SiO2原料[17-18],如图2所示。另外,在实际配料过程中也难以确保1 mol SiO2颗粒恰好与3 mol C粉均匀接触,导致局部原料配比失衡,从而粉体含有残留的C和生成SiC杂相。

图1 SiO2-C-N2体系在不同温度和分压下的优势区域图(采用HSC 6.0 热力学分析软件计算)Fig.1 Predominance area diagram of the SiO2-C-N2 system under different temperature and partial pressure(calculated by HSC 6.0 thermodynamic analysis software)
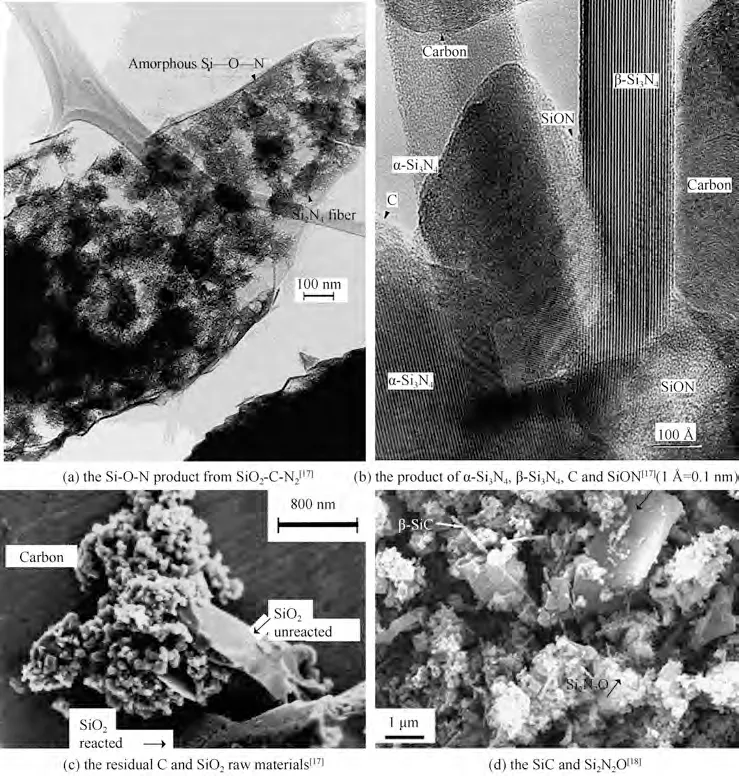
图2 SiO2-C-N2体系生成产物图Fig.2 Product of the SiO2-C-N2 system
从以上分析可知,强化传质是改善粉体质量的关键。当前强化传质主要有以下两种方法。一是破碎外壳提供传质通道。例如,在20 世纪80 年代,Ishii 等[19]将初步氮化的微米级SiO2粉体通过机械破碎暴露出还未被氮化的Si2N2O 和残留的SiO2,然后再进一步氮化。尽管该方法可以消除Si2N2O 和SiO2杂相(XRD 谱图中不再呈现其衍射峰),但是粉体中仍然含有SiC 和C 杂相[20]。二是细化SiO2原料和改善C 的分布状态。最典型的代表是碳包覆纳米SiO2的氮化法。例如,在液相中混合Si源溶液(正硅酸乙酯、硅酸、聚硅氮烷等)和有机碳源(蔗糖、葡萄糖等),并加入添加剂(甘氨酸、尿素、硝酸铵),在一定条件下热解喷雾制备成C 包覆纳米SiO2的复合粉体,然后再氮化合成Si3N4粉体[21-22]。这种方法改善了C 分布均匀性的同时也缩短了扩散传质路径,可消除SiO2和Si2N2O 杂相,但粉体中仍然含有SiC 和C 杂相。为除去粉体中的SiC,1989年Schonfelder等[23]开发出了氯化净化法,即在800~1050℃采用Cl2将固相SiC 转变为SiCl4(g)和CCl4(g),如式(9)所示。为消除粉体中残留的游离C杂质,Ishii等[19]开发出氧化除碳法,即采用O2、CO2或NH3将粉体中的固体C 转变为CO2(g)、CO(g)或CH4(g),如式(10)~式(12)所示,结合氯化净化法成功地将粉体中的C 含量降低到约0.12%(质量),满足了高质量粉体对C含量的要求。但进一步降低C 杂质的可能性不大,因为这部分微量的C杂质既不是SiC也不是游离C,而是存在于晶格中的C杂质。

碳热氮化法已成功实现商业化生产,据企业官网、销售等渠道了解,具有百吨级生产线的国内外企业主要有日本东芝、日本住友化学、福建臻璟新材料科技有限公司和衡阳凯新特种材料科技有限公司,各企业销售的粉体质量如表1 所示。由表中数据可知,α 相、C 含量以及金属杂质都满足高质量粉体的要求,但粉体中的O 含量相对较高。这主要是由于Si 与O 的结合力比Si 与N的结合能力强(Si—O 键能435 kJ/mol,Si—N 键能310~330 kJ/mol),SiO2粉体中的Si—O 键难以完全被Si—N 键置换,导致部分O 残留在晶格中。因此,通常难以获得O 含量小于0.9%(质量)的粉体。这类粉体可用于制备对热导率要求不高的结构陶瓷或光伏领域的脱模剂,难以用于制备高热导率陶瓷,因为晶格中的氧杂质会散射声子,降低热导率。

表1 国内外采用碳热氮化SiO2生产Si3N4粉体质量Table 1 The quality of Si3N4 powders synthesized by carbothermal nitriding of SiO2 in the domestic and overseas
1.2 Si粉直接氮化法
Si 粉直接氮化法,即Si粉与N2反应生成Si3N4粉体,化学反应如式(13)所示。该反应合成Si3N4的路径相对简单,不涉及生成杂相的副反应。由于该反应为强放热反应(在1200℃每生成1 mol Si3N4可释放约822.5 kJ 热量),在实际的反应过程中Si(s)表面的温度要远高于反应设置的温度,从而反应界面处的Si 会液化和汽化形成Si(l)和Si(g),与此同时,Si(l)和Si(g)也都能被氮化生成Si3N4,如式(14)和式(15)所示。在整个反应过程中,部分Si(g)的释放或Si 空位的集聚会在Si 颗粒的内部产生气孔[24],同时Si 颗粒的表面会形成Si3N4外壳[图3(a)][24],并且Si 颗粒间会产生烧结熔聚[图3(b)][24]。

图3 氮化反应过程示意图[24]Fig.3 Schematic diagram of the process of nitridation reaction[24]
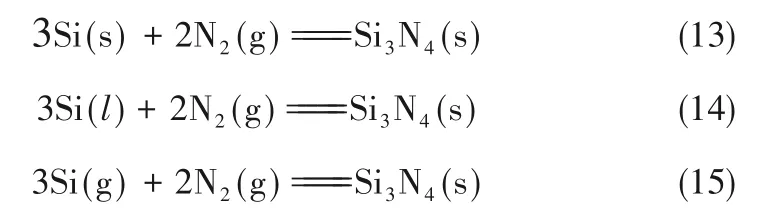
关于固、液、气三相共存的氮化反应和形成Si3N4外壳已形成共识,但其反应机理的认识(是否形成传质障碍层或氮化硅外壳是否会剥落)差异较大。根据文献调研,已经报道的氮化机理主要有以下三种:一是形核和侧向生长[25-27],适用于氮化粒径约100 μm 的Si 粉;二是形核和择优生长[28-30],适用于氮化约0.2 μm 的Si 粉;三是外壳裂纹诱发的表面剥落氮化[31-34],适用于氮化粒径约2.0 μm 的Si 粉。其中氮化粗粉的传质阻力远大于氮化细粉体的传质阻力。Koike 等[35]、Li 等[36]、Jennings 等[32]认为Si3N4外壳是否产生裂纹并脱落与Si 颗粒大小、Si3N4外壳厚度以及Si 与Si3N4的晶格取向密切相关。表2 给出了Si 和Si3N4不同取向时晶格失配应变[35]。由于不规则的Si 颗粒会暴露各种不同的晶面,从而会产生不同的晶格失配应变,也就会导致不同的临界脱落厚度[37],如图4 所示。通过理论建模分析和实验发现[32,35-36],在氮化平均颗粒尺寸约为2.0 μm 的Si 颗粒时,Si3N4外壳脱落的临界厚度为20~300 nm,部分应变较小的位置,诱发裂纹较难,外壳较难脱落,部分应变较大的位置,外壳较容易脱落。

图4 硅粉直接氮化产品TEM图Fig.4 TEM image of product by the direct nitridation of Si powder

表2 Si和α-Si3N4不同取向时产生的应变[35]Table 2 Misfit strain between Si and α-Si3N4 with different orientation[35]
根据其强放热的特性,工业中主要采用自蔓延燃烧技术制备Si3N4粉体,其制备流程为将Si粉紧密堆积或压制成较致密的柱体(确保热量的传递),然后点火引发氮化反应并利用其放出的热量诱发后续氮化反应,见图5(a)、(b)。该技术最显著的优势是节能和经济,只需提供初始点燃热量,后续反应自发进行。理论上,采用超细、电子级高纯Si 粉就可以制备出高质量Si3N4粉体。然而,燃烧过程中表观活化能Ea为292~670 kJ/mol[38-40],表明其仍然受制于扩散传质,且自蔓延燃烧过程中温度梯度大(2400~25℃),导致部分Si 粉液化后被氮化,部分Si 粉被包裹在液相中无法被氮化,部分Si 粉只有表面被氮化,部分Si 粉表面产生裂纹被氮化,部分Si 粉汽化后被氮化[41-45],从而获得内含游离Si 的Si3N4块体[图5(c)、(d)]和少量Si3N4细粉体[图5(f)]。从以上现象可知,仅采用Si 粉为原料的自蔓延燃烧技术难以制备高质量Si3N4粉体。其难点不在于减小粉体粒径和降低杂质。因为尽管Si3N4硬度大,难破碎,且破碎过程会引入大量杂质,但采用先进的粉碎技术反复粉碎和酸洗,粉体的粒径和纯度也能达到高质量粉体的要求。其最大的难题在于消除游离Si 和获得高α 相Si3N4粉体。难以避免游离Si 的内因在于合成反应受扩散传质控制,Si 粉难以被完全氮化。难以获得高α 相的内因在于α 相转化为β 相较为容易,且对温度非常敏感。通常1400℃以上α 相就可以缓慢转变为β 相,且不可逆。自蔓延燃烧合成过程中较大的温度梯度导致粉体中α-Si3N4含量不可控,通常小于70%,极端条件下α 相仅有约1%。因此,如何控制热量和强化传质是自蔓延燃烧技术合成高质量粉体的关键。

图5 自蔓延燃烧反应合成氮化硅反应示意图及产品形貌图Fig.5 Schematic diagram and micrograph of Si3N4 production through self-propagating combustion method
当前,为改善粉体质量已经开发出的强化传质方法主要有以下两种:一是高压氮化法,即提升反应容器中N2的压力(3~10 MPa),为N2内扩散提供更大的驱动力[37,46]。通常只有在N2压力大于3 MPa 时自蔓延燃烧才能顺利进行[47-48],且当N2压力从约5 MPa 提升到8 MPa 时,游离硅的含量从12.0%(质量)降低到3.0%(质量)[46]。此外,常压下Si3N4约在1900℃开始分解,而10 MPa 下Si3N4的分解温度约为2430℃,增大N2压力有利于提高Si3N4收率。然而,N2压力越大,反应越彻底,放出的热量也就越多,粉体中的α 相含量就越低。二是细化粒径和硅粉表面构造裂纹强化传质。最常用的方法为机械活化法[49-54],即在惰性气氛中将粗Si 粉研磨成细粉,缩短了传质距离,同时研磨后细Si 粉表面存在较多微裂纹和缺陷,也为氮气提供了传质通道,可显著降低游离Si 的含量。由于文献中检测手段不同,报道残留的游离硅含量也不同。部分文献报道可彻底消除游离Si,但部分文献认为无法消除游离硅。造成这种差异的主要原因是分析表征手段不同。认为可消除游离Si的判断依据是XRD 中未检测出Si 的衍射峰,这显然不太合理。因为通常当第二相含量低于5.0%(质量)时XRD 无法检测出第二相。相比较而言,采用化学分析的表征手段更为合理,认为其中游离硅的含量为0.11%~0.5%(质量)。当然,如果不计成本无限循环破碎和氮化,理论上也能彻底消除游离Si。
此外,通过控制热量来调控α 相含量也主要有两种方法:一是添加热量“稀释剂”,不仅可以减少液相Si,抑制粗大块体的形成,而且可以降低燃烧合成温度,提升α 相的含量。已开发出的热量“稀释剂”主要有三类,第一类为α-Si3N4粉体,也是最常用的稀释剂[55-56]。通过减少单位体积的放热量来减少热量,同时,α-Si3N4粉体还能充当晶种作用,强化气相反应的形核和生长。由于放热仍然较难控制,温度也难以精确控制在1400℃以下,部分α-Si3N4稀释剂会转化为β-Si3N4粉体。第二类为NaCl、MgCl2、NH4Cl、NH4F 等稀释剂[57-58],利用其熔化、汽化、分解来耗散热量[41-43,51,59],同时,NH4Cl和NH4F等分解产生的HCl和HF 还能与Si反应生成SiCl4或SiF4,而SiCl4或SiF4又会与分解产生的NH3氮化生成Si3N4细粉或晶须。这类稀释剂通常需要复杂的后处理才能较彻底除去Cl、F、Na 和Mg 杂质。第三类为SiO2和C的复合稀释剂[60-61],利用SiO2-C-N2体系吸收热量降低温度,同时也利用碳热氮化合成氮化硅粉体。基于以上强化传质传热的自蔓延技术已成功实现商业化生产Si3N4粉体。表3给出了国内外具有百吨级生产线的主要企业和各自粉体质量指标。值得一提的是,每个企业都有多种型号的粉体,例如Höganäs(H. C. Starck)有4 种粉体,ALZChem 有6 种粉体,烟台同立高科有9种粉体,表中给出的是各企业最优的粉体。现阶段这些粉体主要用于光伏领域和部分先进结构陶瓷。根据中国产业研究院统计,2019 年我国光伏级氮化硅行业市场规模约为10.25 亿元,主要被烟台同立高科、ALZChem、日本UBE 和德国H.C.Starck 占有。从表3 中可以看出,尽管各企业制备的粉体α 相含量较高,但都小于95%,且仍然含有微量的游离Si,难以用于制备高热导率和高强度陶瓷基板。

表3 国内外采用自蔓延燃烧技术生产Si3N4粉体质量Table 3 The quality of Si3N4 powders synthesized by self-propagating combustion technology in the domestic and overseas
尽管自蔓延燃烧技术相对简单且节能,但是难以连续化生产。由于流化床具有传热传质效率高,且可以实现连续批量化生产的优势,最近几十年有大量的研究者尝试采用流化床直接氮化Si 粉合成Si3N4粉体。早在1987 年日本信越化学工业株式会社(Shin-Etsu Chemical Co., Ltd.)就申请了流态化直接氮化法制备Si3N4粉体的专利[60]。在随后的十几年该公司不断优化流化床结构和氮化工艺[61-64],包括两级流化床氮化[61],移动床[62],在流化床中构造急剧温度变化,利用Si 和Si3N4热膨胀系数差异来加速外壳的脱落[63],以及开发连续进出料的氮化工艺[64],逐渐将游离Si 的含量从约30.0%(质量)降低到约1.0%(质量),α 相含量逐渐从约75.0%(质量)提升到96.9%(质量)。但是低游离Si 的指标和高α 相含量不可兼得。例如,α 相含量为96.9%(质量)时,游离Si 却含有约5.2%(质量),游离Si 为1.0%(质量)时,α 相含量却为75.0%(质量)[63]。此外,在连续氮化工艺中,最优粉体中α 相约为93.5%(质量),游离Si 约为2.4%(质量)[64],不满足高质量粉体的要求。除日本信越化学外,众多研究者[39,44,65-70]也在流化床反应器结构优化、热振荡氮化、Fe 或Ca 催化氮化、粒径和气氛优化等方面进行了研究,获得的结论与日本信越化学研究结果一致。在低温下氮化可获得α 相大于95.0%(质量)的Si3N4,游离Si 不可避免,约1.0%(质量)。其主要原因还是传质障碍和热量未得到精确控制。此外,由于超细粉难流化、停留时间难控制、氮化时间长、游离Si 难消除等问题,导致流态化氮化技术进展较缓慢。国内外尚未见商业化生产的报道。最近,魏飞等[71]采用Fe 杂质作为催化剂氮化超细Si 粉,通过造粒在流化床中实现了宏量化制备α-Si3N4纳米线,Si 粉的氮化率约98.9%(质量)(游离Si 1.1%(质量)),α 相约94.6%(质量),纳米线的收率为3.1%(质量)。
1.3 化学气相合成法
化学气相合成Si3N4粉体的反应原理为气相硅源,例如SiCl4、SiH4、SiHCl3、SiBr4等与NH3或N2和H2反应直接生成Si3N4粉体[72-74]。不同反应体系合成Si3N4粉体的质量也不同。SiCl4-N2-H2/NH3和SiHCl3-N2-H2/NH3体系常用于制备Si3N4涂层,直接制备Si3N4粉体难度较大,常需要高能等离子体辅助来合成粉体。由于等离子体火炬的温度高且温度梯度非常大,导致粉体主要为β 相Si3N4粉体。此外,等离子体设备成本较高,产能较小,常见于实验室研究,尚未见工业化制备Si3N4粉体的报道。相比较而言,SiH4-NH3体系更容易合成粉体,且不含Cl 杂质,如式(16)所示。美国通用电气公司早在1978 年就尝试基于该体系采用流化床来合成Si3N4粉体[75],发现在600~1100℃可合成比表面积为5.0~20.0 m2/g的非晶Si3N4粉体,但其中含有约1.0%(质量)游离Si,且没研究其晶化行为。最近,姚奎鸿等[76-77]发现即使对该工艺进一步优化也难以避免游离Si。这主要是由于SiH4不稳定,当温度高于500℃时,部分SiH4就开始分解产生SiH2和游离Si 杂质[77],如式(17)和式(18)所示。SiH4、SiH2、Si 都可以被氮化,且还会形成Si-N-H 的化合物[78],导致氮化反应路径非常复杂。部分SiH4会在氮化反应之前或出气口降温段发生分解,部分SiH4和中间产物会在反应区同时形成Si 和Si3N4。为精确调控反应区的温度,研究者们尝试采用激光局部加热SiH4和NH3来合成Si3N4粉体[79-80]。例如,采用CO2激光局部加热SiH4和NH3合成了约0.5 μm 的无定形Si3N4粉体[80]。但由于SiH4和NH3吸收激光能量存在差异,导致分解和氮化反应仍然不匹配,其中仍然含有约2.0%(质量)的游离Si。若要消除游离Si 首先需要明确游离Si 的存在状态。因为在气相沉积过程中,如果多个气相反应同时生成固相晶核,其粉体结构可能会有三种类型:核壳结构,“枣糕”结构和独立结构。其结构的差异取决于各自的形核和生长速率。如果游离Si 和Si3N4独立存在,那么合成的超细粉体经过二次氮化处理,理论上可消除游离Si。如果是核壳结构或“枣糕”结构,那么消除游离硅有一定难度。因此,后续研究应首先厘清Si 和Si3N4的存在状态。此外,SiH4毒性较大,易燃易爆,安全级别要求非常高。由于以上因素,尚未见化学气相合成Si3N4粉体的工业化生产报道。

1.4 硅胺前体高温热分解法
硅胺前体高温热分解法的反应原理是SiCl4和NH3首先在低温(-80~100℃)合成硅胺前体Si(NH2)4或Si(NH)2,然后硅胺前体在1400~1600℃晶化合成Si3N4粉体,如式(19)和式(20)所示。

该反应原理可追溯到1938年,其中合成前体的反应非常迅速,且放出巨大热量。能否合成高质量Si3N4粉体关键在于如何控制前体的合成反应并分离出高纯前体。根据原料状态的分类,可采用以下六种反应路径低温制备硅胺前体:(1)固相SiCl4与固相NH3反应;(2)气相SiCl4和固相NH3反应;(3)固相SiCl4和气相NH3反应;(4)液相SiCl4与液相NH3反应;(5)液相SiCl4与气相NH3反应;(6)气相SiCl4和气相NH3反应。由于前体合成反应放热量大,维持反应路径(1)~(3)中固相物质冰点的成本非常高,且操作流程较复杂,仅适用于实验室基础研究,不适于工业规模化生产。相比较而言,反应路径(4)~(6)操作相对较简单。国际上[81-84]对反应路径(4)和(5)开展了大量研究,其中最具代表性的是以下两种低温液相合成法:一是低温液相界面合成法[81,84],即低温下SiCl4液体溶解在有机溶剂中并与液氨混合,由于液氨和有机溶剂不互溶而产生分层,因此合成前体的反应在两相界面中进行,产生的副产物NH4Cl溶解在液氨中,不仅解决了反应速率快难控制的难题,也解决了难以分离出高纯前体的难题。该前体经过高温晶化后可获得α 相>95%,BET 约为6 m2/g,且Cl<0.01%(质量)的Si3N4粉体。由于加入了有机溶剂,产品中的C 含量较高[约1.2%(质量)],且大部分C 存在于晶格中,降低C杂质含量难度大。二是低温液氨合成法[85],即在低温(-60~0℃)高压(0.2~10 MPa)的条件下,SiCl4气体通过鼓泡的方式进入高压液氨容器合成硅胺前体,副产物NH4Cl 溶解在液氨中,然后经过多次液氨清洗,过滤,最后分解和晶化前体合成Si3N4粉体,见图6(a)。日本宇部采用该方法制备了全球最好的高质量等轴Si3N4粉体[图6(b)]。但液氨合成前体的反应条件非常苛刻,难以连续化批量运行,必须间歇生产,导致粉体产量小,效率低。此外,硅胺前体具有极强的吸湿性,间歇生产过程中吸湿防护成本非常高。以上两个因素导致该方法制备的高质量氮化硅粉体价格非常高(在80 万~100 万元/吨之间波动)。根据Freedonia 公司统计,氮化硅粉体成本占据了陶瓷成本的1/3~1/2,严重制约了Si3N4粉体应用范围。

图6 日本宇部液氨法制备氮化硅产品流程及产品图Fig.6 Flow chart and microscopic image for preparing high quality Si3N4 powder by UBE industry
在以上六种反应路径中,低温气相合成前体反应条件最温和,最有希望实现连续化批量生产。早在20 世纪80 年代,Clarke[86]尝试采用低温气相合成的前体并通过高温分解和晶化制备Si3N4相粉体。尽管成功合成了粒径约0.3 μm、α相大于95%的粉体,但粉体中Cl 杂质含量较高,即使在1750℃长时间热处理也难以将粉体中Cl降低到0.01%(质量)[87-88]。在粉体烧结过程中,Cl杂质会抑制α 相向β 相的转变,从而难以致密化烧结,同时Cl杂质会散射声子,降低热导率[89-91]。张克鋐等[92-93]也发现了类似的现象。但迄今为止,尚未见降低Cl杂质的有效手段。此外,粉体形貌难以调控,同时存在不规则粉体、等轴粉体以及针状粉体。造成Cl杂质难除和形貌难控的主要原因是对以下几个基础问题的认识还不够。(1)合成温度是否会改变硅胺前体的化学组成还不清楚。首先,合成硅胺前体的热力学数据缺失,难以进行热力学分析。其次,SiCl4和NH3在低温下的反应非常复杂,至今还没有很好认识,仅有少量的文献推演出在-10℃以下合成的前体为Si(NH2)4或Si(NH)2,缺少直接证据。质谱仪分析发现,在约500℃以上时,前体中还会含有(Cl2SiNH)x、(Si2(NH)3Cl2)x、Si3N4(H,Cl)x、(ClSiN)x等物质[94-95],但是-10~500℃之间,前体的化学组成是否随温度而变化还不清楚,难以优选反应区间和调控前体的合成反应。(2)Cl 杂质在粉体中存在的状态及演化行为尚不明确。理论上采用加热分解氯化铵便可除去氯化铵副产物,但是已有研究结果表明热分解法难以脱除Cl杂质。因此,需要明确热分解过程中Cl 杂质存在形式(Si—Cl,N—Cl,H—Cl或NH4Cl)以及Cl杂质在前体、非晶、晶化颗粒中的分布状态(表面、内部、孤立点或连续团簇),才能为设计脱氯方案提供指导。(3)晶化过程中粉体形貌演变机制尚未厘清。尽管部分文献提出了晶须的固-液-气的生长机制,但是晶化过程中同批次粉体在相同条件下形成不同形貌粉体,其形核、生长本征过程和杂质的诱导机制尚不清楚,从而难以精确合成单一形貌的粉体。
2 结论和展望
高质量粉体是制备高性能氮化硅陶瓷的基础,低成本的粉体制备技术是氮化硅陶瓷广泛应用的关键。自20 世纪30 年代起,通过设计反应体系和采用强化传热或传质等手段,氮化硅粉体质量得到了显著的改善。当前低成本制备高质量氮化硅粉体仍面临较大挑战。SiO2碳热氮化法、Si 粉直接氮化法和硅胺前体转化法各自存在的共性难点和可能的解决方案如下。
(1)SiO2碳热氮化法制备粉体的成本较低,但难以合成低氧含量的高质量氮化硅粉体,其共性难点在于固相反应中Si—O键难以完全被Si—N键置换。针对该问题,可进一步优化碳热氮化法的反应路径,将气相SiO 的氮化路径分离出来,并采用气相碳源将其首先转化为Si(g),然后再氮化为Si3N4,有望降低或消除晶格O杂质。
(2)Si粉直接氮化法合成粉体效率较高,成本相对较低,但难以合成高α相含量且不含游离Si的氮化硅粉体,其共性难点在于强化固相传质和精确控制热量。针对该问题,可借助原位高温透射电镜等现代分析技术,深入研究不同粒径、不同形貌以及暴露不同晶面Si粉的氮化行为,结合界面反应和异质界面应变及传质理论模拟分析,进一步完善Si粉直接氮化法的机理,明确有利于强化传质的特殊晶面,优选出具有特定粒径和形貌Si粉原料,并调控化学气相沉积合成Si粉的形核与生长动力学条件,精准批量合成满足特殊要求的Si粉,同时开发流态化氮化技术,利用流化床传热、传质效率高的优势,将反应界面温度控制在1400℃之下,则有望消除游离Si,合成纯α相粉体。
(3)硅胺前体转化法可合成高质量的氮化硅粉体,但成本高,其共性难点在于前体合成反应的精准调控和制备工艺中吸湿防护成本的控制及连续化生产。针对该问题,可将传热传质效率高且易于批量化生产的流化床技术与硅胺前体转化法相结合,设计可连续化运行的气相或液相合成新工艺,提高生产效率和产量,将是未来低成本制备高质量氮化硅粉体的重要发展方向。
随着粉体制备技术的迭代和产业结构的升级,综合性能优异的氮化硅陶瓷可在冶金、化学、机械、电子等领域获得广泛的应用,例如在航空航天领域有望用于制备发动机的陶瓷叶片、涡轮外环、增压器转子、锒块、挺柱、摇臂锒块、电热塞等高温零部件;在电子通信领域有望取代传统Al2O3和AlN 陶瓷基板;在机械工程领域有望进一步拓展在陶瓷刀具、轴承、球阀、密封环、喷嘴等关键部件的应用范围,预计未来几年内氮化硅将会迎来高速发展时期。
