基于BSA-seq 的黄褐棉纤维长度候选基因发掘
2022-01-13王议萍周婉婷朱心慧于梓璇李馨蕊汪保华
陈 奇,王议萍,陈 艳,周婉婷,汤 备,朱心慧,于梓璇,李馨蕊,汪保华
(南通大学 生命科学学院,江苏 南通 226019)
棉花是世界上重要的经济作物,也是最重要的天然纤维作物和纺织工业原料。纤维长度是评价纤维品质的重要指标,在一定范围内,纱线强度随着纤维长度增加而增大;此外,相较于短纤维,长纤维的加工效率更高,并能生产出质量更高的细纱线[1]。唐淑荣等[2-3]研究指出,我国的栽培棉品种还存在一些突出的问题,包括:断裂比强度普遍偏低,纤维长度、强度和细度搭配不合理,不能满足棉纺织工业纺纱工艺的用棉需求,我国栽培棉的纤维品质需进一步提升。
棉花纤维长度是由多基因控制的数量性状,因此,定位其在染色体上的具体区域对后续候选基因的进一步发掘具有重要意义。经过多年研究,国内外学者鉴定了很多棉花纤维长度数量性状基因座(quantitative trait locus,QTL),例如:在棉花1 号染色体上发现的一个稳定的纤维长度QTL,在不同研究团队的不同遗传背景下都能检测到。Chee 等[4]利用海岛棉与陆地棉种间回交高世代群体定位到Chr.1染色体上的纤维长度QTL(qFL-chr1),海岛棉等位基因在3 个不同的回交高世代家系衍生的群体中均可提高纤维长度,解释12%~24%的表型方差。Shen 等[5]利用BC3F2植株构建了3 个独立的近等导入系群体,进一步验证了该QTL;其中一个近等基因系R01-40-08 具有94.3%的轮回亲本基因组,能显著提高纤维长度。Xu 等[6]利用R01-40-08 构建了目标区间分离的大群体,精细定位该QTL 至0.9 cM区间,并筛选到两个候选基因GOBAR07705、GOBAR25992。除Chr.1 染色体上的稳定主效QTL 外,研究人员也多次在Chr.7 染色体[7-9]和Chr.25 染色体[10-12]上检测到纤维长度QTL。此外,Ning 等[13]检测到Chr.21 染色体上一个稳定的纤维长度QTL(qFLD11-1),在7 个环境下均能检测到。Wang 等[14]利用一套陆地棉重组自交系群体鉴定到13 个纤维长度QTL;其中,在Chr.18 染色体上检测到纤维长度QTL(qFL18.1)位于分子标记PGML1145~JESPR178区间。虽然目前已鉴定了很多纤维长度QTL,但克隆候选基因进而得到应用的并不多,因此,有必要进一步发掘纤维长度候选基因并验证其功能,最终用于棉花分子育种实践。
棉属分为A、B、C、D、E、F、G、K 8 个染色体组,共52 个种,其中包括45 个二倍体和7 个异源四倍体棉种[15]。在野生棉资源中,四倍体野生棉与陆地棉倍性相同,便于直接杂交获得稳定遗传的后代;前人研究利用陆地棉与毛棉、陆地棉与达尔文棉种间群体,都鉴定到一些稳定的纤维长度QTL[16-17]。在棉花7 个异源四倍体里,黄褐棉与陆地棉遗传距离最远[18],因此,最有可能存在新的基因位点,挖掘并利用这些新的基因对拓宽陆地棉遗传背景和改良纤维品质具有重要的科学意义和应用价值。例如:Shen等[19]将陆地棉与黄褐棉杂交并回交,构建了包含71个家系的染色体片段代换系群体,定位到29 个纤维品质QTL,并发现部分QTL 的有利基因来自于黄褐棉。
本团队前期研究利用黄褐棉与陆地棉种间F2群体构建了遗传图谱,在此基础上构建了高世代回交群体BC3F2、BC3F2:3和BC3F2:4,共鉴定出131 个控制纤维品质性状的QTL[20-23]。基于回交高世代群体选择了一套导入系,其中一些导入系纤维品质表现突出;对这套导入系开展了多年重复的田间试验,对纤维品质性状开展了QTL 定位,共定位到15 个纤维长度QTL,其中包括Chr.18 染色体上一个纤维长度主效QTL(qUHM-18-1),其增效基因来自黄褐棉,解释23.36%的表型方差,加性效应为1.42 mm[24],并由此通过分子标记辅助选育出一个带有qUHM-18-1且纤维长度突出的黄褐棉导入系IL9(纤维长度3 年平均值为33.3 mm)。本研究拟利用IL9 与其陆地棉轮回亲本PD94042 构建的F2大群体,通过极端性状混池测序分析(bulked-segregant analysis sequencing,BSA-seq)结合QTL 定位结果,进一步挖掘黄褐棉纤维长度候选功能基因,为棉花分子育种提供基因资源。
1 实验材料与方法
1.1 植物材料
本实验室前期分子辅助选择了一个优质黄褐棉导入系IL9,在IL9 的基因组构成中,黄褐棉导入片段约占15.3%[24]。利用IL9 和陆地棉轮回亲本PD94042 杂交并自交,构建得到F2代次级群体,共1 244 个单株,开展纤维品质表型鉴定。
1.2 BSA-seq 分析
1.2.1 材料准备
盛花期按单株采集BC3F2群体的棉花叶片放置于-80 ℃超低温冰箱保存,在获得纤维品质表型数据后,根据测序要求挑选纤维长度最长和最短的棉花材料的叶片各30 份,再选取相同数量的亲本材料PD94042 和IL9 的叶片,提取基因组DNA 组成4个混池,由北京组学生物科技有限公司进行基因组重测序,测序深度为30×,参考基因组为棉花全基因组序列(http://mascotton.njau.edu.cn/Data.htm)。
1.2.2 文库构建及测序
首先,检测样品基因组的DNA,合格的DNA 用超声波打断的方法将其片段化;然后,进行片段的纯化和末端修复,进而开展3′端加A 和测序接头的连接;之后,利用琼脂糖凝胶电泳选择片段大小,进行PCR 扩增形成测序文库。构建好的文库先进行质检,再利用Illumina HiSeq 平台对质检合格的文库进行测序。
1.2.3 测序结果质控
高通量测序得到的原始测序序列(Raw data 或Raw reads)包含序列信息及其对应的测序质量信息,必须经过质控进而得到Clean data 后才能用于后续分析。质控方法包括:低质量数据过滤、碱基测序质量分布和碱基类型分布。
1.2.4 候选基因的发掘与鉴定
将质控后得到的Clean data 重新比对到参考基因组上,进而开展后续的变异分析,根据比对结果进行插入缺失(insertion-deletion,InDel)和单核苷酸多态性(single nucleotide polymorphism,SNP)的检测及注释。对于InDel 和SNP 的检测主要使用GATK软件工具包来实现,主要检测过程可参考GATK 官方网站的BestPractice(https://www.broadinstitute.org/gatk/guide/best-practices.php),随后使用一种高效的软件工具ANNOVAR 对SNP 进行注释。对于差异基因的关联分析,本实验通过两种方法来确定候选区域,分别为欧式距离(euclidean distance,ED)算法关联分析和基于BSA-seq 的SNP-index 算法关联分析。SNP 质量分布和SNP 突变频谱被用来检测所得SNP 数据的可靠性,最终根据SNP-index 算法计算与性状关联的候选区域。SNP-index 算法如下:基于基因分型的结果,筛选亲本中纯合的且亲本间具有多态性的SNP 位点。将其中的一个亲本作为参考,计算两个子代在亲本间标记位点的SNP-index(SNP频率),即统计混池和亲本(或参考基因组)在某一个碱基位点上相同或不同的reads 数,进而计算出不同reads 数占总reads 数的比例,即为该碱基位点的SNP-index。完全与其相同的SNP-index 为0;完全与其不同的SNP-index 为1。计算Δ(SNP-index),即两个子代的SNP-index 之差:Δ(SNP-index)=SNP-index(极端性状B)-SNP-index(极端性状A)。该差值越接近于1,则标记SNP 与目标性状之间的关联度越大,把超过阈值线的区域定为候选区域。对分析所得的候选基因区域内的基因进行基因同源性分析及相关功能深度注释,以此来筛选所要得到的候选基因。
2 结果与分析
2.1 表型结果统计分析
利用本实验室前期分子辅助选择的优质黄褐棉导入系IL9 和陆地棉轮回亲本PD94042 杂交构建得到F2代次级群体,共1 244 个单株,在相同条件下,对亲本材料PD94042、IL9 和F2代群体的纤维长度表型数据进行测量和记录。该部分表型数据被用来进行BSA 分析所需数据的选择,F2代次级群体平均表型结果如表1 所示。从表中的数据可以看出,相较于亲本PD94042,导入系IL9 明显具有更优良的纤维表型表现,后代群体的纤维长度得到了改良。
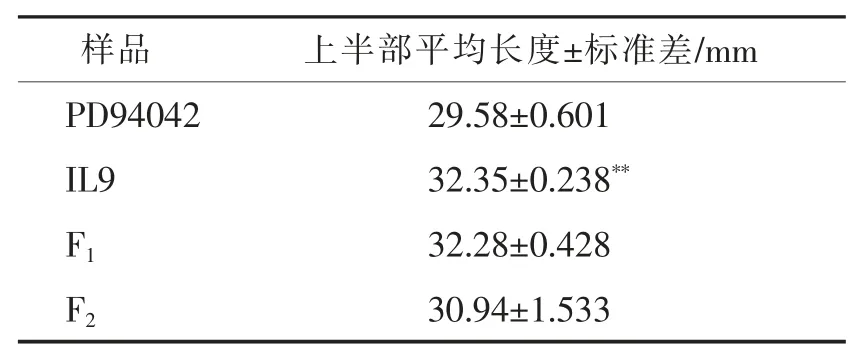
表1 F2 代次级群体纤维长度表型数据Tab.1 Phenotypic data of fiber length in the F2 population
2.2 与参考基因组的比对统计
将重测序获得的reads 重新比对到参考基因组上,进而开展后续的变异分析。本研究的测序样品序列reads 总数在277~327 Mbp 之间,GC 碱基数都在35%左右,GC 分布正常,而且各个样品Q30 的值都在90%左右,说明过滤后的结果较为可靠。
经过过滤和修饰读取后,4 个样本reads 比对到参考基因组上的覆盖率都超过98%,双端测序序列均比对到参考基因组上,且距离符合测序片段的长度分布均超过94%。对于两个极端性状DNA 混池,测序深度达到33×,且参考基因组中至少有10 个碱基覆盖的位点占基因组的百分比均超过81%,每个样品的测序数据信息充分,测序质量合格。结果表明该数据符合突变分析检测的标准,可成功用于Illumina 测序分析。
2.3 混池SNP 检测统计
样品表型之间的差异往往是其基因组序列上的变异所导致的,在BSA-seq 分析中,利用GATK软件工具包,把重心放在检测样品间序列的SNP上。SNP 指由基因组核苷酸水平上的变异引起的DNA 序列多态性,包括由单碱基的转换、颠换和单碱基的插入与缺失等引起的同义突变和错义突变。通过与参考基因组的对比,得到每一个样本和参考基因组的SNP 数目,以及样本之间的结果比较。混池间的SNP 结果统计如表2 所示。
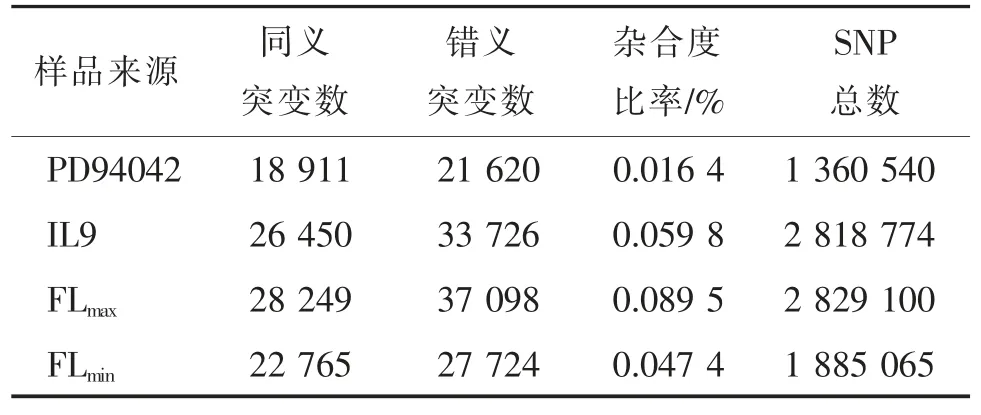
表2 混池SNP 结果统计表Tab.2 SNP information of different samples
从表2 中可以看出,FLmax中的SNP 总数高达2 829 100,而FLmin中也包含1 885 065 个SNP。为进一步统计和可视化4 个样本间的SNP 重复性,我们做了SNP 的统计结果Venn 图(图1)。从Venn 图整体分布情况来看,FLmax混池基因组更接近于IL9,FLmin混池基因序列则更接近于PD94042。这也从侧面说明了子代中控制更长纤维长度的基因大部分来源于黄褐棉导入系IL9,该结果为本研究课题的展开和后续工作提供了理论支持。

图1 纤维长度混池样品SNP 统计Venn 图Fig.1 Venn diagram of fiber length SNPs
2.4 候选基因区域筛选
本研究通过基于BSA-seq 的SNP-index 关联分析来计算两个混池间等位基因的基因型频率,进而获得与目标性状相关联的基因组区域。根据SNP-index 方法关联阈值判定,首先计算两个子代在亲本间标记位点的SNP 的频率(即SNP-index),计算Δ(SNP-index),即两个子代的SNP-index 作差,超过阈值线的为候选基因组区域。当拟合后的Δ(SNP-index)置信度为0.99 时,共得到3 个与棉花纤维长度性状相关联的染色体区域,分布于Chr.7、Chr.10、Chr.18 3 条染色体上(图2)。

图2 纤维长度混池SNP-index 关联分析Fig.2 SNP-index association analysis of fiber length
通过对关联分析所得到的3 个候选区域的进一步统计,包括这些区域所处的染色体位置(起始和结束位点)、序列长度和候选基因数目,得到候选区域的相关信息统计(表3)。从表中数据可以看出,纤维长度性状候选基因主要集中在Chr.7、Chr.10、Chr.18 3 条染色体的3 个基因区域,涉及的基因数分别为7、21、6 个。
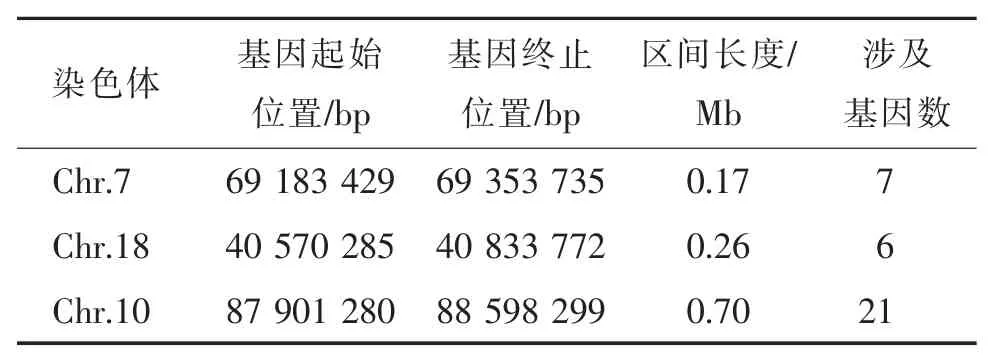
表3 纤维长度基因候选区域相关信息统计表Tab.3 Statistics of candidate gene regions related to fiber length
2.5 候选基因筛选
在前期研究中,我们鉴定了一个位于18 号染色体上的纤维长度主效QTL(qUHM-18-1),候选区域覆盖染色体的31 696 068~51 883 215 bp 基因组区间,而本次BSA-seq 研究中鉴定到的Chr.18(D13)候选基因区域与该QTL 的染色体区域有部分重叠,且重叠区域包含了引起氨基酸变化的单核苷酸变化,因此我们选取Chr.18 染色体作为后续验证的主要候选基因区域。我们对BSA 候选区域内的基因开展了进一步的整理,随后通过GO 功能注释信息和KEGG 通路注释信息,进而对区域内的候选基因进行与纤维长度性状相关的筛选。综合QTL 及BSA-seq 结果,该区域存在6 个相关基因,分别为Gh_D13G1292、Gh_D13G1293、Gh_D13G1294、Gh_D 13G1295、Gh_D13G1296、Gh_D13G1297。通过GO功能注释信息和KEGG 通路分析,最终筛选到3 个候选基因:Gh_D13G1294、Gh_D13G1295 和Gh_D 13G1296(基因序列信息见表4)。
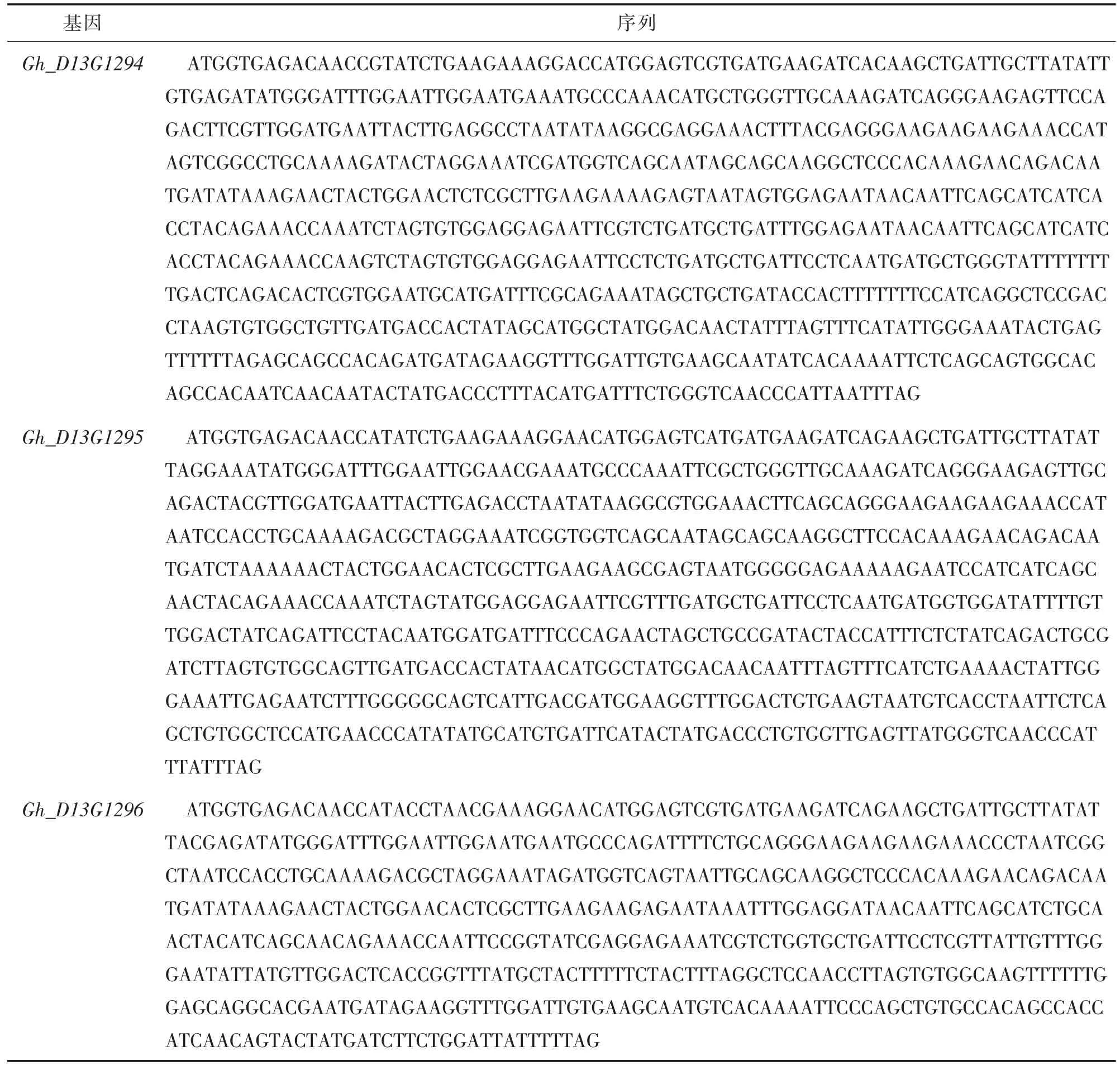
表4 目的基因序列信息Tab.4 Sequences of the target genes
3 结论
前期研究中,我们基于一套黄褐棉导入系群体开展了纤维品质QTL 定位,鉴定了15 个纤维长度主效QTL[24]。选择纤维品质突出的导入系IL9 与其陆地棉轮回亲本PD94042 杂交并自交,构建了F2大群体,结合表型鉴定结果,开展了纤维长度的BSA-seq 分析。基于SNP-index 和ED 的关联算法,控制纤维长度的数量性状位点被分别定位在Chr.7、Chr.10、Chr.18 共3 条染色体上3 个相关区域内,涉及的基因数分别为7、21、6 个。前期基于黄褐棉导入系的QTL 定位研究鉴定到Chr.18 上一个纤维长度QTL(qUHM-18-1),其增效基因来自黄褐棉,解释23.36%的表型方差[24]。该QTL 是在前人研究发现的来自达尔文棉、毛棉等四倍体野生棉种之外,增效基因来自黄褐棉的新的纤维长度主效QTL,为陆地棉纤维品质改良提供了新的基因资源。通过比较该QTL 和BSA-seq 候选区域的位置信息,我们将候选基因区域缩小到更精细的区域。对分析所得区域内的候选基因进行基因同源性分析及相关功能注释筛选,最终筛选出最可能和纤维长度的生长发育建成相关的3 个候选基因,为Gh_D13G1294、Gh_D 13G1295 和Gh_D13G1296。
Gh_D13G1294 编码MYB-like102(MYB102),在拟南芥中,MYB102 转录因子可以通过调节乙烯生物合成[25]和细胞分裂素水平[26]来促进植物体的生长发育,但在棉花中的分子作用机制尚不明确。Gh_D13G1295 编码myb domain protein 4r1 转录因子,属于R2R3-MYB 转录因子基因家族,是植物中最大的转录因子基因家族之一,该基因家族在植物的生长发育建成和代谢调节方面发挥广泛作用。Gh_D13G1296 编 码myb domain protein 16 转录因子(MYB16),该转录因子在植物体的表皮形成[27]和细胞形态变化发生[28]中发挥着重要作用。在分子功能方面,这三个基因都是蛋白质编码基因,且都与染色质结合(GO:0003682)相关,通过编码相关蛋白与染色质、蛋白质、DNA 的纤维网络或者在染色体间期组成真核细胞核染色体的RNA 进行选择性和非共价相互作用完成相关功能。KEGG 通路分析发现这几个基因都主要集中作用在DNA 的结合过程中,以及作用于其合成蛋白质的转录因子方面。这些基因可能在棉花纤维长度的发育过程中起重要作用,在后续研究中,将进一步验证其功能,最终用于棉花分子育种实践。
