标准曲线法、标准加入法和标准物质法测定磷矿石中氧化镁含量对比
2022-01-10张江坤黄永丽叶罕章
史 鑫,张江坤,张 慧,姜 威,黄永丽,叶罕章
(国家磷资源开发利用工程技术研究中心,云南昆明 650600)
磷矿石作为磷化工行业的主要原料,其品质直接影响化工产品的生产。磷矿石中杂质MgO 含量对湿法磷酸及磷铵生产有着较大的影响,其含量越高,混合料浆的黏度越大,料浆浓缩就越困难,容易形成不溶化合物[1]。磷矿石中MgO 含量已成为酸法加工评价磷矿质量的主要指标之一。测定磷矿石中MgO 含量的方法主要有火焰原子吸收光谱法(测定范围w(MgO)0.1%~10.0%)和沉淀分离-乙二胺四乙酸(EDTA)容量法(测定范围w(MgO)>0.5%)[1-2]。传统化学分析方法,滴定终点主要以肉眼判断,终点颜色不易观察,容易造成较大误差,导致测定结果不准确。笔者采用电感耦合等离子体发射光谱仪,使用3种不同方法测定磷矿石中的MgO 含量,特别是针对MgO 含量高的磷矿石样品,通过比较得出最佳方法。
1 实验部分
1.1 仪器及工作参数
ICPA7400 型全谱直读等离子体发射光谱仪(美国ThermoFisher(赛默飞世尔)公司),配置玻璃雾化器和雾化室,ITEVA操作软件。仪器最佳工作参数如下:发射功率1 180 W,冷却器流量12 L/min,辅助气流量0.5 L/min,雾化器压力0.2 MPa,垂直观测高度12 mm,蠕动泵转速75 r/min,样品提升量1.5 mL/min,样品冲洗时间30 s,短波积分时间7 s,长波积分时间5 s。
1.2 主要试剂
HNO3、HCl均为优级纯;镁标准储备溶液(钢铁研究总院),ρ(MgO)1 000 mg/L;实验用水为超纯水,20 ℃电阻率为18 MΩ·cm;高纯氩气,纯度大于99.99%。
1.3 实验方法
1.3.1 样品的前处理
准确称取磷矿试样0.10 g(精确至0.000 1 g),置于300 mL 烧杯中,加少量水润湿试样,加入HCl-HNO3溶液(体积比3∶1)20 mL 混匀,在200 ℃电热板上加热,待烧杯中液体蒸发至近干时取下烧杯,冷却后将液体转移至100 mL 容量瓶中,用水定容,过滤后待测。同时做空白实验。
1.3.2 标准溶液系列的配制
标准曲线法:分别取1 000 mg/L的镁标准储备溶液0、2.5、5.0、10.0、15.0、20.0 mL 置于6 个100 mL 容量瓶中,加入HCl 溶液5 mL,用水定容,配制成MgO 质量浓度分别为0、25、50、100、150、200 mg/L的标准溶液系列。
标准加入法:配制MgO 质量浓度分别为0、25、50、100、150、200 mg/L的标准系列溶液;称取试样标准物质(GBW 07210)0.1 g(精确至0.000 1 g),置于250 mL烧杯中,按王水溶矿法处理,冷却后移入标准系列溶液中,该标准系列MgO质量浓度分别为0、29.3、54.3、104.3、154.3、204.3 mg/L。
标准物质法:取混标物质(w(MgO)8.19%)2.442 4 g(精确至0.000 1 g),置于250 mL 烧杯中,按王水溶矿法处理,冷却后定容至1 000 mL容量瓶中,配制成质量浓度为200.0 mg/L的氧化镁标准溶液,过滤后逐级稀释成质量浓度为0、25、50、100、150、200 mg/L的标准溶液系列。
2 结果与讨论
2.1 分析谱线的选择
将电感耦合等离子体发射光谱仪点燃稳定0.5 h 后,依次引入蒸馏水和镁标准溶液,通过ITEVA软件的光谱仪谱线库在镁的分析谱线进行扫描,以谱线信噪比大、干扰小及检出限低等为依据选择镁的分析谱线,选择波长285.2 nm 为Mg 的最佳分析谱线。
2.2 工作曲线
将配制好的标准溶液在仪器工作条件下进行测定,绘制标准曲线。在仪器最佳工作条件下,测定空白溶液11次,以测定结果的标准偏差的3倍作为方法检出限,结果见表1。
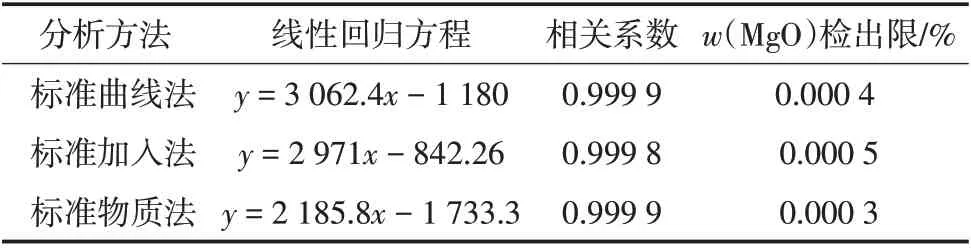
表1 线性参数及检出限
由表1 可知,3 种方法得到的线性回归方程相关系数均≥0.999 8,满足测定要求,检出限分别为0.000 4%、0.000 5%、0.000 3%。
2.3 共存元素干扰
磷矿石中含大量的P、Fe、Al、Si、Ca,为了考察上述离子的干扰,按实验方法,配制Mg 质量浓度为10.00、20.00 mg/L,而P、Fe、Al、Si、Ca质量浓度均为1 g/L 的混合标准溶液,按照实验方法测定混合标准溶液中待测元素的质量浓度,结果见表2。
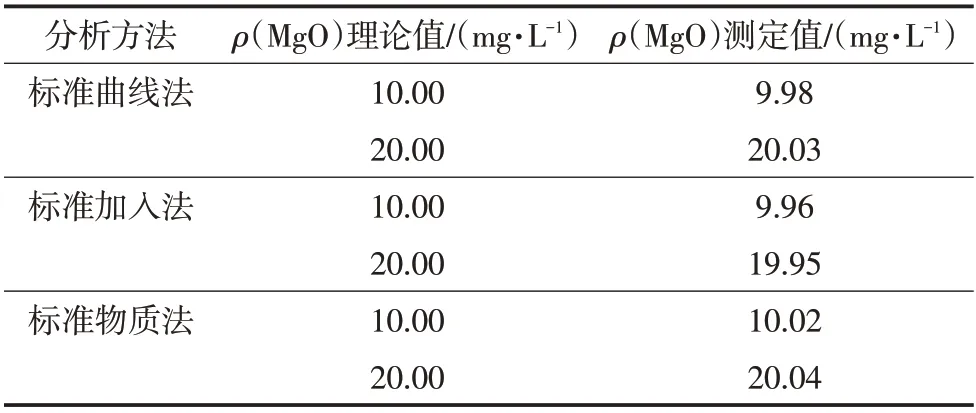
表2 不同干扰元素共存下待测液的测定结果
由表2 可知,磷矿石中P、Fe、Al、Si、Ca 的存在对MgO的测定没有干扰。
2.4 加标回收率实验
按照实验方法测定磷矿石样品,对样品GBW07210、GBW07211、X-1 进行加标回收率实验,结果见表3。
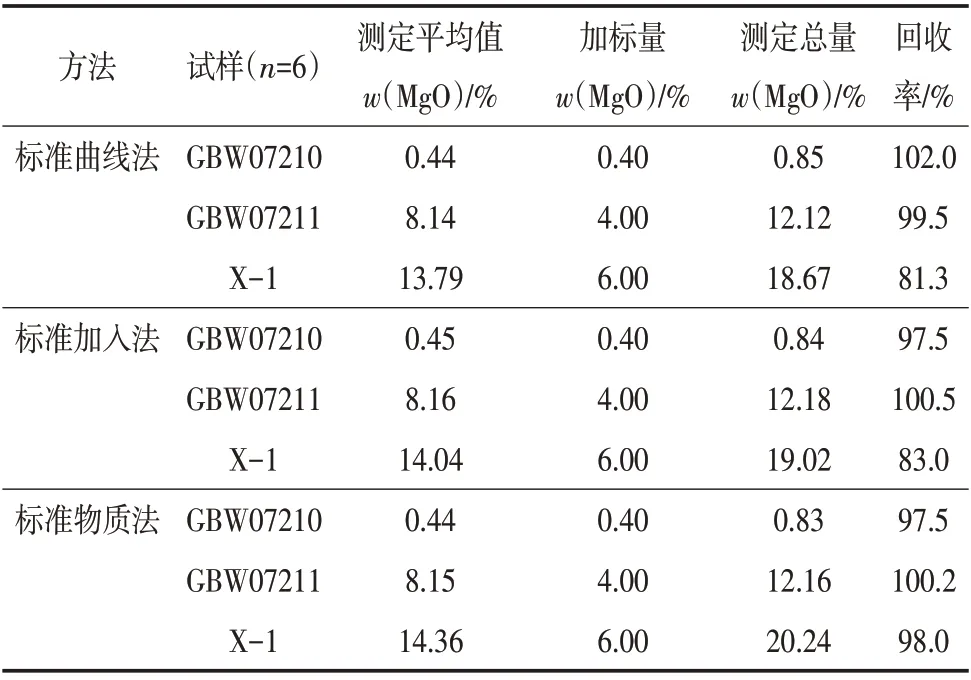
表3 3种方法的加标回收率实验结果
从表3可以看出:标准曲线法测定结果的加标回收率为81.3%~102.0%,标准加入法测定结果的回收率为83.0%~100.5%,标准物质法测定的结果的加标回收率为97.5%~100.2%。测定低浓度样品时,3种方法的准确度均满足测试要求,测定高浓度样品时,仅标准物质法能满足要求。
2.5 样品分析结果对比
分别使用3种测定方法对不同的磷矿石样品进行分析,测定结果见表4。
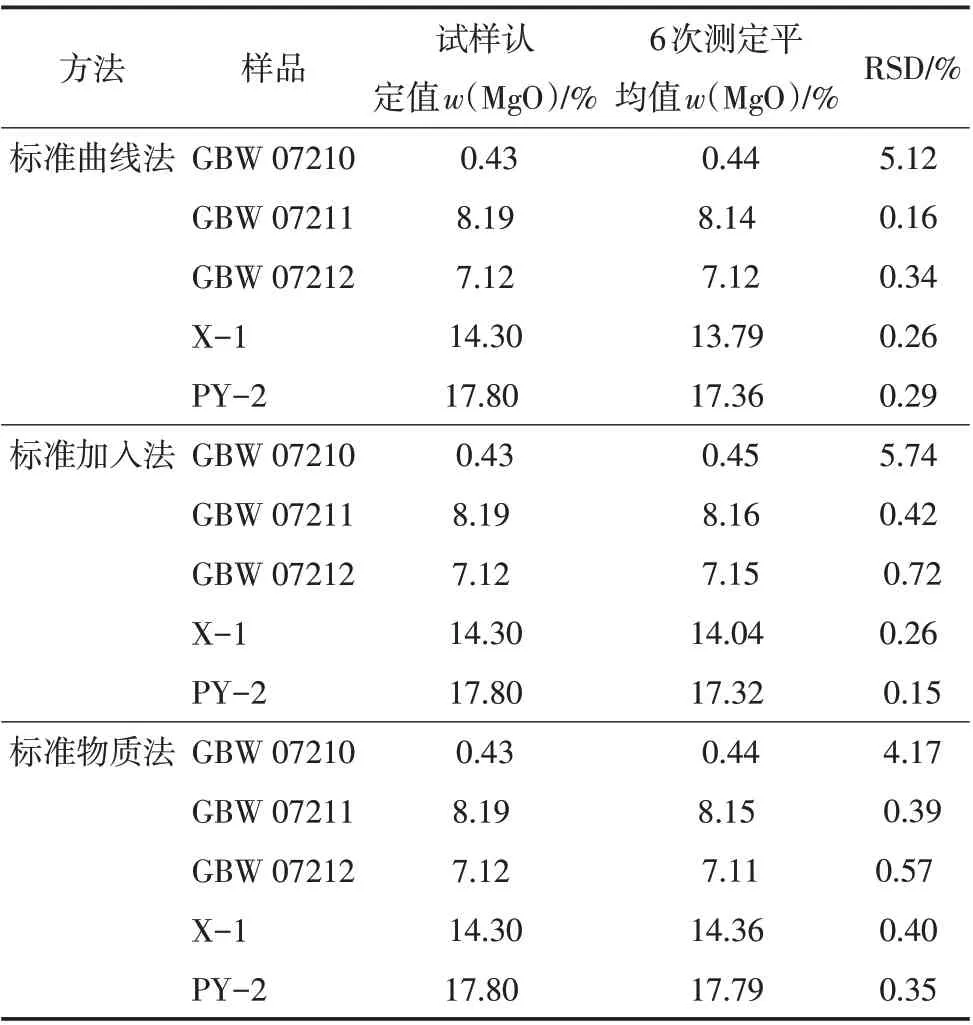
表4 3种方法分析结果对比
从表4 可以看出,标准曲线法的RSD(n=6)为0.16%~5.12%,标准加入法的RSD 为0.15%~5.74%,标准物质法的RSD 为0.35%~4.17%,均满足测定要求。测定氧化镁含量较低的磷矿石样品时,3种方法测得的结果都在测定误差允许范围之内,满足测定要求。但对于氧化镁含量较高的磷矿石样品,标准曲线法和标准加入法测定的结果都低于认定值,超出误差允许范围(w(MgO)>5%,允许差为0.30%),只有标准物质法测定的结果在测定误差允许范围之内,准确度较高。
3 结论
总体来看,标准曲线法、标准加入法和标准物质法3种方法测定的线性都较好,均可测定氧化镁含量较低的磷矿石样品;对于氧化镁含量较高的磷矿石样品,只有标准物质法测定的结果在允许误差范围之内,准确度较高。因此,对于氧化镁含量较低的磷矿石,标准曲线法、标准加入法和标准物质法的测定效果相当,可以选择其中任意一种方法进行测定,但考虑到检测效率、准确性,更推荐标准物质法;对于氧化镁含量较高的磷矿石,标准物质法具有可以消除复杂样品中基体干扰的优势,准确度、精密度更高,更符合测定要求。
