小麦miRNA启动子的基因组分析
2022-01-08任治鹏王多佳娄贵成王政委
任治鹏,王多佳,田 宇,娄贵成,李 畅,王政委,张 达,苍 晶
(东北农业大学生命科学学院,黑龙江哈尔滨 150030)
MicroRNA(miRNA)是一类长度为21~24 nt的非编码RNA,广泛存在于植物中,通过负调控其靶基因,参与调控植物的生长发育和逆境胁迫响应[1-2]。miRNA的生物合成过程主要包括miRNA基因的转录、初始转录本加工为成熟miRNA以及成熟miRNA装载形成RNA诱导的沉默复合体(RNA-induced silencing complex,RISC)[3-7]。RISC通过酶切降解靶基因mRNA或者抑制靶基因mRNA的翻译,从而对靶基因进行转录后水平上的调控[8]。
在植物中,能够转录形成miRNA的miRNA基因大部分位于基因间隔区,作为独立的转录单位,只有部分miRNA基因位于蛋白质编码基因内,能与宿主基因共同转录[9]。研究表明,miRNA基因由RNA聚合酶Ⅱ(RNA polymerase Ⅱ,Pol Ⅱ)转录[10]。Pol Ⅱ型启动子包括核心启动子区和上游作用元件,核心启动子区主要由TATA-box、转录起始位点(transcription start site,TSS)等构成[11]。了解miRNA基因的位置、启动子的TSS、特定顺式作用元件等上游序列特征,对于研究miRNA的表达模式及miRNA介导的调控网络具有重要意义[12]。近年来,通过生物信息学分析结合高通量测序,对植物miRNA基因的启动子开展了一定的研究。如在拟南芥中,Megraw等[13]和Xie等[14]通过5′-RACE的方法,发现大部分拟南芥miRNA启动子包含TATA-box。Zhou等[15]通过CoVote的方法,在拟南芥、水稻等植物中鉴定了基因间miRNA基因的启动子,结果表明,miRNA基因与蛋白质编码基因均由Pol Ⅱ型启动子启动,并具有特定的上游元件。Zhao等[16]利用cDNA数据对水稻和拟南芥两种植物miRNA启动子元件进行比较,同时通过ChIP方法对拟南芥miRNA基因的TSS进行了预测[17]。随着植物基因组研究的发展,促进了miRNA启动子的鉴定和研究。如Cui等[18]通过基因组数据,定位了水稻miRNA前体(miRNA precursor,pre-miRNA)在染色体上的位置,并通过TSSP软件预测了miRNA基因的TSS、TATA-box等核心启动子区。Liu等[19]和Han等[20]利用大豆基因组数据对miRNA基因的启动子特征进行了相关分析。Kanjanawattanawong[21]等发现,橡胶树中对乙烯响应的miRNA启动子具有多种植物激素相关作用元件。Zhou等[22]利用TSSP-TCM软件对拟南芥、毛果杨、水稻、高粱4种植物的miRNA启动子进行生物信息学分析,发现基因间和基因内以及保守和非保守miRNA的启动子具有不同的基因组分布特征及特异性作用元件。此外,研究者在拟南芥[23]、水稻[24]miRNA启动子中也发现具有与胁迫相关的特异性转录因子结合元件。
六倍体(2n=6x=42,AABBDD)普通小麦(TriticumaestivumL.)是全球种植最广泛的农作物之一,为人类提供了20%的消耗能量[25]。目前,对小麦miRNA的研究主要集中于克隆鉴定、表达特征分析以及通过预测靶基因进行功能研究等方面[25-26],然而关于小麦miRNA启动子的研究报道较少。近年来,国际小麦基因组测序联盟(International Wheat Genome Sequencing Consortium,IWGSC)对中国春小麦基因组的组装工作已经完成,其公布的小麦全基因组序列信息对于小麦miRNA启动子的分析研究具有极大的促进作用。本研究通过生物信息学方法对miRNA基因组位置分布、miRNA启动子预测以及顺式作用元件的富集和特异性进行研究,以期在基因组水平对小麦miRNA启动子有一个较为全面的了解,为小麦miRNA的转录调控探究以及新miRNA的预测提供依据。
1 材料与方法
1.1 小麦miRNA基因位置的预测
所有的小麦miRNA序列来源于miRBase数据库(Release 22.1,http://www.mirbase.org/)[27]。从Ensembl Plants(ftp://ftp.ensemblgenomes.org/pub/plants/release-48/fasta/triticum_aestivum/dna/)下载中国春小麦基因组序列信息。使用URGI BLAST(https://urgi.versailles.inrae.fr/blast/?dbgroup=wheat_iwgsc_refseq_v2_chromosomes&program=blastn)[28]进行小麦pre-miRNA的基因组定位,选择identities=100%的blast结果作为miRNA基因的位置,对于identities≠100%的miRNA则将identities≥97%且mismatches≤2的结果作为miRNA基因的位置[19]。预测的miRNA基因通过Mapchart 2.30[29]软件进行小麦染色体图谱的绘制。所有能够定位于小麦基因组上的miRNA基因根据两种方法进行分类,第一种分类方法是根据miRNA保守性分为保守和非保守miRNA基因,鉴定方法如下:首先利用miRBase提供的所有物种pre-miRNA序列建立本地blast库,然后将所有小麦pre-miRNA进行本地blast比对。如果其他植物中存在identities>85%且alignment length>90%的相似序列[22],则该基因为小麦保守miRNA基因,否则为非保守性miRNA基因。第二种分类方法则根据miRNA基因在染色体上的位置进行分类,通过JBrowse(https://urgi.versailles.inra.fr/jbrowseiwgsc/gmod_jbrowse/)[30]判断miRNA基因的分布情况,将miRNA基因分为基因间和基因内两种类型。基因间miRNA位于蛋白质编码基因之间,而基因内miRNA序列位置则与蛋白质编码基因重叠[15]。判断miRNA基因染色体位置参考的编码蛋白质基因数据为IWGSC中国春Annotation v1.1数据库[31],包括可高信度(HC)和低信度(LC)蛋白质编码基因座。
1.2 小麦miRNA基因启动子的预测
首先通过Zhou等[15]的方法获得pre-miRNA的基因间5′端上游序列,当pre-miRNA与上游蛋白质编码基因转录方向相同时,如果它们之间的距离大于2 400 bp,则检索pre-miRNA上游2 000 bp序列;如果距离小于2 400 bp,则检索上游蛋白质编码基因下游400 bp与pre-miRNA之间的序列。当pre-miRNA及其上游蛋白质编码基因转录方向相反时,如果它们之间的距离大于4 000 bp,则获取pre-miRNA上游的2 000 bp序列,如果距离小于4 000 bp,则检索从pre-miRNA到中间点(上游蛋白质编码基因与pre-miRNA之间)的序列。将以上方法获得的序列作为潜在的启动子预测区域,利用TSSP(http://www.softberry.com)进行小麦miRNA启动子及TSS的预测。
1.3 小麦miRNA基因启动子上游顺式作用元件的分析
利用PlantCARE数据库(http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)[32]对miRNA启动子TSS到上游2 000 bp序列中的顺式作用元件进行分析。对于有多个启动子的miRNA基因,为获得尽可能多的顺式作用元件信息,选择距离pre-miRNA起始位点最近的TSS进行分析。为了进一步研究miRNA启动子区域基序的特异性,通过MEME(https://meme-suite.org/meme//tools/meme)[33]对miRNA启动子上游序列中长度为10 bp的基序进行鉴定,选择结果中前20个基序进行分析,其他设定为默认值。利用全基因组蒙特卡罗模拟方法获得基序的Z-score,从而判断各基序在小麦miRNA启动子的特异性[15],具体方法如下:首先将所有获得的miRNA启动子序列作为目标集,然后在小麦基因组上随机选择长度为2 000 bp的序列作为参考集,参考集与目标集的序列数目相同;通过FIMO(https://meme-suite.org/meme//tools/fimo)统计特定基序在目标集和参考集miRNA序列上平均数量,分别记为Nt和Nr。Z-score的计算公式为Z=(Nt/Nr)=σ,它能测量目标集中的基序平均出现次数与参考集样本的均值之间的归一化差异[22]。利用CpGPlot(http://emboss.bioinformatics.nl/cgi-bin/emboss/cpgplot)对小麦miRNA TSS上游序列中的CpG岛进行分析。
2 结果与分析
2.1 小麦miRNA基因的染色体定位
目前为止,miRBase数据库(Release 22.1)共收录122个小麦pre-miRNA序列。小麦pre-miRNA序列和中国春基因组序列blast结果表明,105个(86.1%)pre-miRNA定位于小麦染色体上的150个基因座上,而其余17个(13.9%)pre-miRNA位于未知染色体或基因组的基因座上,下文中不对此类pre-miRNA进行统计。pre-miRNA分布在小麦所有42条染色体上,其中A组染色体上有54个,B染色体上有56个,而D组染色体上有40个,93个(76.2%)pre-miRNA在染色体上只有1个拷贝,含有2个和2个以上拷贝的pre-miRNA分别有4和8个,共占比 9.84%,其他17个pre-miRNA的拷贝为0。
2.2 小麦miRNA基因的启动子预测结果
150个小麦miRNA基因座中有148个能够获得启动子潜在区域,对148个miRNA基因座5’上游序列进行启动子预测,由于部分miRNA基因座能够预测到多个启动子,因此共获得166个miRNA潜在启动子。115个(77.7%)小麦pre-miRNA基因能够预测到一个启动子,其中,69个基因的上游序列只能预测到一个启动子,而其他基因具有多个启动子。
TSS是重要的启动子核心元件,对小麦miRNA基因TSS位点与pre-miRNA距离分布进行统计分析,发现大部分小麦miRNA基因的TSS分布在上游0.8 kb区域内以及1.0~1.6 kb区域内,占全部启动子TSS数的81.9%(0~0.8 kb:54.2%,1.0~1.6 kb:27.7%)。在所有上游区域中,小麦miRNA基因的TSS在上游0.2 kb区域内分布最多(24.1%),而在上游0.8~1.0 kb区域分布较少(5.4%)。
根据miRNA在基因组的位置不同,可分为基因间miRNA和基因内miRNA,从图1A可以看出,两种miRNA的TSS均在基因上游0.2 kb区域内分布较多,不同的是基因内miRNA在上游0.2~0.4 kb、0.6~0.8 kb、1.4~1.6 kb间也具有较多的TSS分布,而基因间miRNA在这几个区域内无明显的分布特殊性。根据miRNA的保守性,可分为保守性miRNA和非保守性miRNA,从图1B可以看出,两种miRNA的TSS均在基因上游0.2 kb区域内分布最多,而与非保守miRNA相比,保守miRNA在上游1.4~1.6 kb区域内也具有较多分布。
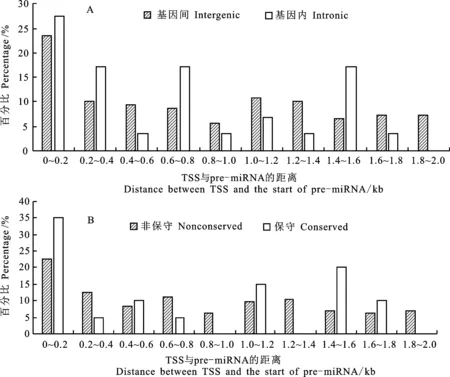
A:基因间和基因内miRNA TSS的分布百分比;B:非保守和保守miRNA TSS的分布百分比。
2.3 小麦miRNA基因启动子上游的特异性顺式作用元件
利用PlantCARE对所有miRNA基因TSS上游2 000 bp序列进行顺式作用元件分析,结果(图2)表明,miRNA启动子区域中含有的三种顺式作用元件较多,分别为CAAT-box、TATA-box和Unnamed_4。此外与ABA响应相关的元件(ABRE)、与MeJA响应相关的元件(TGACG-motif、CGTCA-motif和MYC)、与光响应相关的元件(G-box)以及与多种胁迫和代谢调控相关的元件(MYB)在小麦miRNA基因上游的占比也较高。
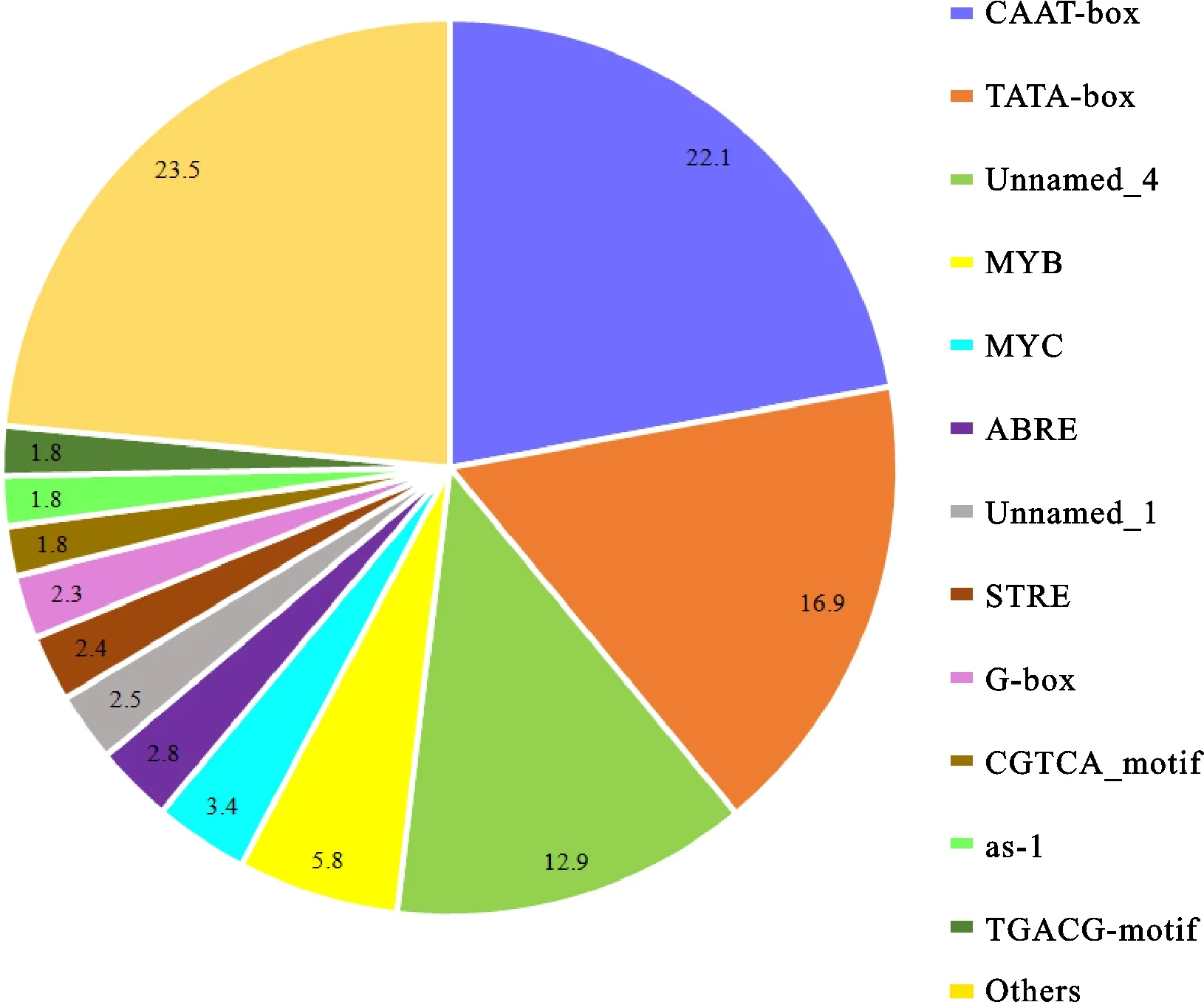
图中数据为启动子上游顺式作用元件所占百分比。
为进一步鉴定小麦miRNA基因启动子上的基序特异性,通过MEME获得在TSS上游序列出现频率较高的且长度为10 bp的基序,然后利用全基因组的蒙特卡罗模拟计算获得基序的Z-score。Z-score的大小在一定程度上能反应基序在miRNA启动子上的特异性,Z-score大于2的基序具有miRNA基因启动子特异性,与miRNA的转录调控有关的可能性较高[22];而Z-score小于2的基序在其他基因组区域普遍存在,因此不作为miRNA启动子重要基序进行研究。根据以上标准,获得了3个Z-score≥2的小麦miRNA启动子特异性基序(表1)。

表1 小麦miRNA基因启动子特异性基序
除顺式作用元件外,CpG岛也是真核生物polⅡ型启动子的重要特征之一。由于本研究中MIR9670和MIE979可能通过同一个启动子进行转录,因此对114个miRNA基因启动子的CpG岛进行分析,CpGPlot预测结果表明, 61.4%的小麦miRNA基因TSS上游序列有CpG岛分布,启动子区域含有1、2、3和4个CpG岛的miRNA基因分别有41、17、10、2个。
3 讨 论
本研究首先将miRBase数据库目前登录的所有小麦pre-miRNA序列定位于小麦基因组上,在122个pre-miRNA中有部分序列无法通过blast获得基因座,其可能原因为:(1)基因位于数据库中的未知染色体上;(2)pre-miRNA的序列信息不完全,或所研究品种的pre-miRNA序列与参考的中国春基因组序列存在差异;(3)由于小麦基因组较大,组装困难,目前提供的基因组版本存在部分染色体序列的缺失。本研究染色体定位结果表明,所有小麦染色体上均存在miRNA基因。前人研究表明,在三个染色体组中,B组染色体上的miRNA基因分布最多,根据IWGSC数据库,编码蛋白的基因也在B组染色体上的分布最多[21]。本研究选择blast结果为100%的染色体位置为miRNA基因座,因此多拷贝的miRNA基因序列相同。具有多拷贝的miRNA基因中,只有MIR6197和MIR9774在三个染色体组上均具有拷贝,其他基因只在一个或两个染色体组上具有拷贝,这说明大部分基因在不同染色体组上存在序列不同的情况。在动物中,miRNA基因通常聚簇并形成多顺反子RNA共转录,而只有部分植物miRNA基因存在成簇miRNA。与非成簇miRNA不同,成簇的多个miRNA可通过同一个启动子进行转录[18]。Singh等[34]研究表明,小麦中209个miRNA存在于89个多顺反子基因座上。本研究中,由于使用的miRBase数据库的注释miRNA信息有限,只发现了1个小麦miRNA簇,该miRNA簇中的MIR9670和MIR9779位于6D染色体上,为非保守miRNA,未在Singh等[35]的研究中报道。对MIR9670和MIR9779的启动子预测结果发现,只有一个miRNA启动子位于上游序列,说明这两个miRNA可能通过同一个启动子进行转录。小麦成簇miRNA的启动子特点可通过其他miRNA库进行更深层次的研究。
150个miRNA基因中有148个具有启动子潜在区域,MIR1133和MIR1135基因与上游蛋白质编码基因距离过近,无法获得启动子区域。本研究通过TSSP对小麦miRNA基因的polⅡ型启动子进行了预测,结果表明,大部分pre-miRNA(77.7%)上游具有潜在的启动子序列,而少部分miRNA基因无法预测到启动子,原因可能为:(1)原始miRNA序列较长,利用pri-RNA加工后形成pre-miRNA序列信息进行启动子预测,其启动子可能位于pre-miRNA的上游2 kb以外;(2)大多数启动子预测软件都使用同源搜索的方法,因此可能无法预测miRNA启动子的非保守性启动子;(3)由于基因组的重复性,部分基因组上具有多个拷贝的pre-miRNA序列为假基因,不发生转录,因此无法进行启动子预测[19]。miRNA根据基因位置分为基因间miRNA和基因内miRNA,基因内miRNA通常与宿主基因共同转录,但Cui等[18]的研究表明,基因内miRNA也可能具有单独的启动子,形成独立的转录本。本研究对基因内miRNA和基因间miRNA启动子数目分别进行了统计,发现74.3%的基因内miRNA至少具有1个polⅡ型启动子。以上结果表明,小麦中相当一部分的基因内miRNA也同样具有独立的启动子,而未预测到启动子的基因内miRNA则可能由宿主基因启动子启动转录。
TSS是基因的转录起始位点,因此TSS的预测对于miRNA基因转录特点的研究具有一定的意义。本研究对TSS与pre-miRNA的距离进行统计分析,结果表明,TSS大多数位于pre-miRNA序列上游800 bp内,尤其上游200 bp内。该结果与大豆和水稻中的miRNA基因TSS统计结果类似[18-19],这说明小麦等植物的大多数miRNA核心启动子区域与pre-miRNA序列比较接近,miRNA的PolⅡ启动子近端区域的核心启动子区可能对miRNA的转录起到更大的作用。
植物基因启动子上的顺式作用元件能够识别结合特定的转录因子,从而对基因的转录进行相应的时空特异性调控[24]。本研究对TSS上游序列进行了顺式作用元件分析,结果表明,miRNA基因启动子区域存在多种顺式作用元件。其中TATA-box最多,TATA-box是一种广泛存在的DNA基序,拟南芥和水稻中miRNA特异性基序的功能尚不清楚,但新发现的基序对于在小麦中鉴定新的特异性miRNA以及进行miRNA的试验分析具有一定的借鉴意义。除顺式作用元件外,CpG岛也是重要的启动子序列特征[35]。本研究在小麦中部分miRNA基因上游序列鉴定出了CpG岛,而前人在拟南芥miRNA启动子中未鉴定到CpG岛,水稻miRNA启动子中鉴定出的CpG岛也较少[15],烟草中的MIR169基因家族中也未鉴定到CpG岛的存在[36]。以上结果说明,小麦、水稻等单子叶植物中,CpG岛的分布情况可能与双子叶植物不同。
miRNA在植物的基因调控中起到了重要作用,启动子是控制miRNA基因表达的重要结构,因此对于miRNA基因启动子的分析研究具有较大的意义。本研究利用公布的小麦基因组信息对小麦miRNA启动子进行了分析,研究了小麦miRNA的染色体定位以及TSS分布、顺式作用元件特异性等启动子特征,相关结果对于小麦miRNA表达调控的研究及新miRNA的预测具有一定的借鉴意义。在未来的研究中,随着小RNA测序以及分子实验技术的进步,可获得更多的miRNA功能信息,并结合试验方法对相关结果进行进一步的验证。
