Production of renewable aromatics and olefins by catalytic co-cracking of fatty acid methyl esters and methanol
2022-01-05LIUSenGUOYuqianSUNPeiyongZHANGShenghongYAOZhilong
LIU Sen , GUO Yu-qian , SUN Pei-yong , ZHANG Sheng-hong,* , YAO Zhi-long
(1.Beijing Key Laboratory of Enze Biomass Fine Chemicals, College of Chemical Engineering, Beijing Institute of Petrochemical Technology, Beijing 102617, China;2.College of Chemistry and Environmental Engineering, Anyang Institute of Technology, Anyang 455000, China)
Abstract: Catalytic cracking of triglycerides and their derivatives (e.g., fatty acid methyl esters, FAMEs) by HZSM-5 zeolite offers a promising route to produce renewable aromatics and olefins, but it is primarily hindered by the rapid catalyst deactivation caused by coke.In this work, the co-cracking of FAMEs and methanol over HZSM-5/Al2O3 composites was developed to regulate the product distribution and slower the catalyst deactivation.Co-feeding methanol with FAMEs enhanced the olefin selectivity at the expense of aromatics, and the total selectivities of aromatics and olefins added up to 70.9% with an optimized methanol content of 60%.The co-feeding of methanol not only promoted the olefin yield but also retarded the consecutive H-elimination of aromatics to polycyclic aromatics, thus reducing the coke formation and prolonging the catalyst lifespan.Under the conditions of 450 °C, 0.16 MPa and a space velocity of FAMEs at 4 h-1, increasing the methanol blending ratio in FAMEs from zero to 50% reduced coke from 17.8% to 10.1% after reaction for 12 h.Besides, the spent catalyst for the co-cracking reaction could be easily regenerated by coke combustion, yielding similar structure, acidity and activity to those of the fresh one.
Key words: catalytic cracking;fatty acid methyl ester;methanol;aromatic;olefin
Light aromatics and olefins are produced mainly via energy-intensive processes such as reforming and cracking of petroleum fractions[1].With the declining petroleum resource and increasing environmental concerns, biomass as an alternative and renewable energy source to produce fuels and chemicals,especially BTX (benzene, toluene, and xylenes) and C2-4olefins, has significantly been encouraged to reduce the environmental footprint of petroleum usage.Different from starch, sugar, and lignocellulose biomass, triglyceride-rich biomass has a similar skeleton structure and physical properties (density,boiling point, hydrogen/carbon ratio,etc.) to those of the petroleum-derived hydrocarbons[2].Triglycerides and their derivatives are, thus, considered as promising feedstock to complement the fossil-based counterparts in the production schemes of current refineries such as cracking and refining[3].Producing fuels and chemicals from edible vegetable oils, however, seems much less viable in China, as the gap between the domestic supply of vegetable oils and their market demand is expanding with its increasing population[4].By contrast,fatty acid methyl esters (FAMEs), which are derived from waste cooking oils via transesterification, provide an alternative feedstock to produce renewable aromatics and olefins.
HZSM-5 zeolite has been widely applied in the catalytic cracking of triglycerides into hydrocarbons like aromatics and olefins, due to its adjustable acidity and unique intersecting pores preferring the formation of monoaromatics over polyaromatics[5-8].The formed aromatics, however, tend to condense and fuse into polyaromatics masking on the external surface of HZSM-5, causing the pore blockage and thereby a rapid deactivation[9].Strategies such as dealumination[10], introducing hierarchical structures[11],and doping with Zn and Ga[12]have been developed to attenuate coking.Despite the promoted catalyst stability, these measures always generate undesired effects on catalyst activity and/or selectivity.Dealumination of HZSM-5 reduces the strong acid sites responsible for cracking and cyclization reactions,thereby leading to inferior activity and aromatic selectivity in the cracking of triglycerides; While the introduction of hierarchical porosity into HZSM-5 increases significantly light olefins at the expense of aromatics.By comparison, doping Zn and Ga with dehydrogenation capacity promotes the transformation of olefins into aromatics.Nevertheless, Ga was found to facilitate the deep dehydrogenation of aromatics to graphitic coke, which contributed to a more pronounced decline in the lifetime of HZSM-5 zeolite[12].It is still a challenge to balance monoaromatic formation and further dehydrogenation to polyaromatic coke precursors by directly modifying HZSM-5 zeolite.
In addition to the upgrading of HZSM-5 zeolite,advanced process technologies such as coupling reactions have been developed to attenuate catalyst coking in biomass cracking.The co-cracking of small alcohol molecules with bio-oils improved the stability of HZSM-5 and the aromatic yield due to the hydrogen transfer between methanol and bio-oils[13-15].Methanol has a hydrogen to carbon effective ratio ((H/C)eff) of 2[16], much higher than the (H/C)effvalue of bio-oils.The surplus hydrogen released from alcohol cracking compensates for the hydrogen required for the cracking of bio-oils with a low (H/C)eff, thus attenuating the catalyst deactivation.Improving the integral (H/C)effratio of feedstock by co-feeding reactants with high(H/C)effratios is, therefore, an alternative strategy to retard the deactivation of HZSM-5 zeolite in the catalytic cracking of biomass[17].
Similarly, introducing methanol to triglyceride would prolong the catalyst lifespan, particularly when the triglyceride and methanol crackings require the same active sites and share similar reaction intermediates and products.The integration of triglyceride cracking and“methanol to hydrocarbon (MTH)” is expected to achieve less coke, as well as a promoted energy efficiency by thermally coupling endothermic cracking of triglycerides and exothermic MTH process.For this purpose, industrial HZSM-5 extrudates bound with boehmite have been prepared and evaluated for the FAMEs and methanol co-cracking in the present work.The effect of methanol blending contents on the cracking of FAMEs was investigated in terms of reaction product distribution and catalyst stability.Besides, a reaction pathway for the co-cracking of FAMEs and methanol was further proposed to elucidate the integrated cracking process.This work aims to develop a practical technology that enables the sustainable production of aromatics and olefins from triglycerides and their derivatives.
1 Experimental
1.1 Materials
FAMEs derived from waste cooking oils were supplied by Jiangxi Xufeng Chemical Co., Ltd.The raw materials consisted mainly of FAMEs with 16 or 18 carbon atoms, which accounted for more than 95%.HZSM-5 zeolite with a Si/Al ratio (n(SiO2)/n(Al2O3))of 25 was purchased from Nankai University Catalyst Co.Ltd., and boehmite was provided by Sinopec Catalyst Co.LTD.
1.2 Catalyst preparation
HZSM-5 zeolite and boehmite were mixed at a mass ratio of 3∶1 in a certain amount of 10% HNO3aqueous solution, followed by extrusion, drying at 120 °C overnight, and calcination at 550 °C in air for 4 h.The formed HZSM-5/Al2O3composites were then cut into extrudates of 2.0-3.0 mm in length and 1.5 mm in diameter before use.
1.3 Catalyst characterization
Nitrogen adsorption-desorption isotherms were measured at -196 °C using an Autosorb-iQ analyzer(Quantachrome).The specific surface area was estimated from nitrogen adsorption data in the relative pressure (p/p0) range from 0.05 to 0.25 using the Brunauer-Emmett-Teller (BET) method, and the total volume was calculated from the adsorbed amount of N2atp/p0=0.99.Thet-plot method was used to determine the micropore volume and the discrimination between the external and internal surface areas of catalysts.Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku D/max2500 diffractometer (CuKα,λ= 0.15418 nm, 40 kV, 40 mA) at a scan rate of 2(°)/min.The morphology of catalysts was examined using scanning electron microscopy (SEM, Shimadzu SSX-550) at an accelerating voltage of 20 kV.The29Si magic-angle spinning nuclear magnetic resonance (29Si MAS NMR) measurement was performed on a 400 MHz solid-state NMR spectrometer (Bruker,Avance Ⅲ) at 79.5 MHz.Carbonaceous species deposited on the used catalyst were quantified by thermogravimetric analysis (TGA) on an SDT-650 simultaneous thermal analyzer (TA Instrument) from room temperature to 800 °C in air.
Temperature-programmed desorption of NH3(NH3-TPD) was carried out on a DAS-7200 TPDanalyzer (Hunan Huasi).Generally, samples (100 mg)were first treated at 500 °C for 1 h and cooled down to 100 °C in He (40 mL/min), followed by exposure to NH3(30 mL/min) for 0.5 h at the same temperature.The NH3-saturated sample was then purged by He at 100 °C until a constant baseline was recorded by the thermal conductivity detector (TCD) connected to the reactor.Finally, the sample was heated to 800 °C at a rate of 10 °C/min, with the desorbed NH3continuously monitored by the TCD.
The pyridine-infrared (Py-FTIR) spectra were measured on a Fourier transform infrared (FT-IR)spectrometer (Nicolet 6700) in the range of 1300 to 1800 cm-1.Before the measurement, the tableting sample was treated at 350 °C under a vacuum for 2 h.After cooling down to 50 °C, pyridine vapor was injected into the sample cell under a nitrogen flow of 30 mL/min, and the adsorption continued for 0.5 h.The sample saturated with pyridine was then heated to 200 °C and evacuated at this temperature for 0.5 h under 10-5torr to remove the weakly adsorbed pyridine.Finally, the temperature was increased to 350 °C to detect the medium and strong acid sites.According to the absorbance measured at around 1540 and 1450 cm-1,the Brønsted (B) and Lewis (L) acid sites were quantified using the molar extinction coefficients reported by Emeis[18].
1.4 Cracking reactions
Cracking reactions were carried out in a fixed-bed reactor (Φ20 mm × 600 mm), as illustrated in Figure 1.The reaction temperature was controlled by a thermometer inserted into the catalyst bed, and the pressure was kept at 0.16 MPa by a back pressure regulator.Calibrated pumps (Laballience Series II) and mass flow controllers (SevenStar D07-11C) were respectively used to feed liquid reactants (FAMEs,water, and methanol) and N2to the reactor.Typically,10 g catalysts were loaded in the center of the reactor and sandwiched by two glass bead (O.D.= 0.5 mm)zones.After treatment by steam (WHSV = 1 h-1, N2=100 mL/min) at 480 °C for 12 h under ambient pressure, the reactor was cooled to 450 °C before feeding FAMEs and methanol.The weight hourly space velocity (WHSV) of FAMEs was generally set at 1 h-1, with the mass ratio of methanol to FAMEs varied from 1∶4 to 2∶1 in case of co-cracking reactions.The gaseous reaction stream exiting from the reactor was quenched to 0 °C before passing through a gas-liquid separator, and the condensed liquid phase was collected every two hours for analysis.

Figure 1 Simplified flow diagram of the setup for co-cracking FAMEs and methanol
1.5 Product analysis
The mixture from the reactor outlet was divided into gas, aqueous, and oil phases after separation.The gas products were composed mainly of light hydrocarbons with 1-6 carbon atoms and COx.Their composition was determined by a Shimadzu gas chromatography (GC) GC-2014 equipped with a capillary column (HP-Al2O3/KCl, 50 m × 0.53 mm ×15 μm) and a flame ionization detector (FID).While the aqueous phase comprising predominantly methanol and water was analyzed using an Agilent GC-7890A equipped with a capillary column (SupercowaxTM10,30 m × 0.53 mm × 1 μm) and an FID.The oil phase contained unreacted FAMEs and liquid hydrocarbon products such asparaffins, olefins, and aromatics.A Shimadzu GC-2010 equipped with a capillary column(HP-5, 30 m × 0.32 mm × 15 μm) was used to quantify the liquid hydrocarbons.The relative response factors used for quantification were obtained from the analysis of liquid hydrocarbon standards of known composition.Besides, PONA (paraffins, olefins, naphthenes, and aromatics) analysis was also performed on an SP-3420A GC with an HP-PONA column (50 m × 0.20 mm ×0.50 μm) to quantify isomers of olefins and aromatics in the liquid hydrocarbons.
The conversions of FAMEs (x(FAMEs)) and methanol (x(CH3OH)) were defined as the mass ratios of the converted reactant to the fed one, as listed in Eq.(1) and (2), respectively.The mass balance (MB),which was calculated from the mass ratio of the total products to the converted FAMEs and methanol (Eq.(3)), varied between 94% and 100% for the studied reactions.While the selectivity of each product(s(CxHy)) was determined with a normalization method due to the satisfactory reaction mass balance within(97 ± 3)%, as seen in Eq.(4).However, neither water nor COxwas considered when calculating selectivities,as they were out of focus in this work.
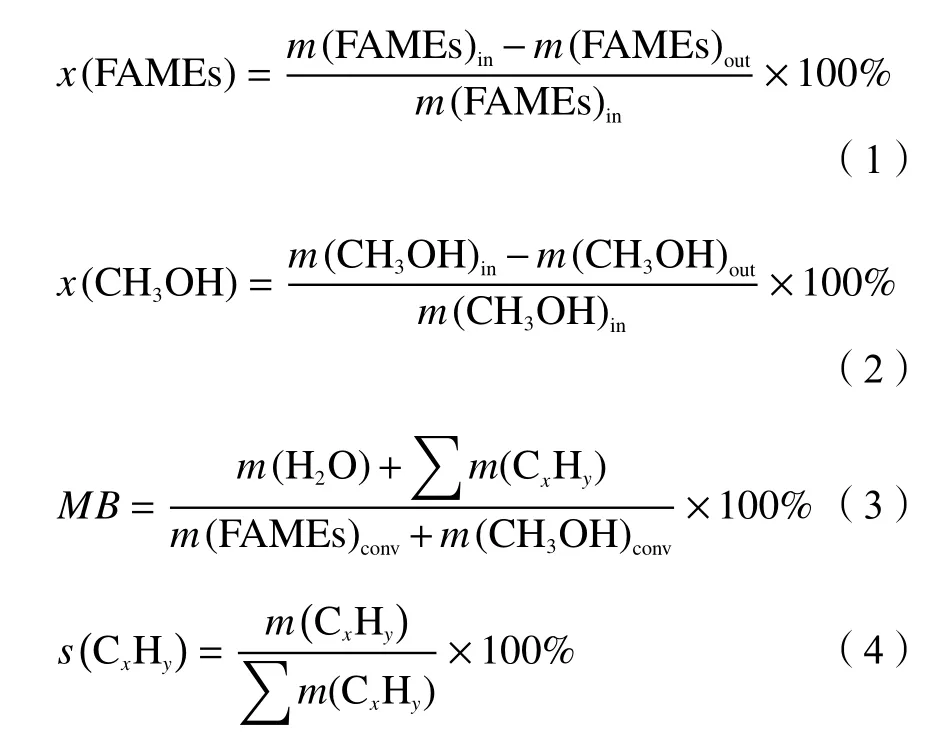
Herein,m(FAMEs)convrepresents the mass of converted FAMEs, equal to the mass difference of FAMEs between flowing in (m(FAMEs)in) and flowing out (m(FAMEs)out).So arem(CH3OH)conv, (m(CH3OH)in),andm(CH3OH)out.
2 Results and discussion
2.1 Catalyst characterization
The HZSM-5/Al2O3catalyst after steam treatment was examined by SEM, XRD, and N2physical adsorption techniques, with results shown in Figure 2 and Table 1.HZSM-5 zeolite consisted of agglomerates of single crystals with uniform sizes ofca.250 nm, while alumina seemed much fluffy with many cavities.Unlike both, the HZSM-5/Al2O3composites were composed of HZSM-5 crystallites dispersed well on the porous alumina (Figure 2(c)).Nevertheless, the catalyst exhibited an identical XRD pattern to HZSM-5, giving a set of typical diffraction peaks in the 2θranges of 8°-10° and 22.5°-25°, which correspond to an MFI framework.This indicates that the MFI structure of HZSM-5 was well maintained after introducing the boehmite binder and the subsequent calcination and steam treatment.Also, the surface area (SBET) and pore volume (vt) of HZSM-5/Al2O3were not significantly affected by steam,decreasing from 345 m2/g and 0.434 mL/g to 305 m2/g and 0.411 mL/g, respectively, see Table 1.Interestingly, the decreases were comparable to the losses in the internal surface area and micropore volume, implying that steam reduced mainly the microporosity of HZSM-5 zeolite.

Figure 2 SEM images of Al2O3 (a), HZSM-5 (b) and HZSM-5/Al2O3 (c); XRD patterns (d) and N2 adsorption-desorption isotherms (e) of the steam-treated HZSM-5/Al2O3 catalyst

Table 1 Physicochemical properties of HZSM-5/Al2O3 catalysts
Besides the microporosity, the steam treatment reduced the acidity of HZSM-5, especially the strong acid sites, as reflected by the NH3-TPD profiles in Figure 3(a).Two NH3desorption peaks appearingaround 190 and 365 °C, which can be respectively assigned to the desorption of NH3adsorbed on the weak and strong acid sites[19], are observable for both bare and steam-treated HZSM-5 zeolites.However, the high-temperature peak is much smaller for the hydrothermally treated sample, suggesting a loss in its strong acid.The loss in the acidity of HZSM-5 was probably caused by the dealumination via steam treatment, as suggested by the29Si NMR spectra of HZSM-5 shown in Figure 3(b).For both calcined and steam-treated zeolites, the resonances at -113.8 and-116.9 could be assigned to the Si (4Si,0Al) sites, and the resonance near -110.8 could be attributed to Si(3Si,1Al) sites; Other resonances below -100 were not identified for either catalyst, indicating the absence of Si (2Si,2Al) sites[20].According to the deconvolution results of29Si NMR spectra and the established method to calculate the Si/Al ratio[21], the estimated (Si/Al)fratio of HZSM-5 zeolite increased from 23 to 31 after the steam treatment, see Table 1.The rising (Si/Al)fratio indicated that some tetrahedrally coordinated Al atoms were substantially removed by steam, in line with the reduced acidity of HZSM-5 measured by NH3-TPD.

Figure 3 Effect of steam treatment on the NH3-TPD profiles (a), 29Si MAS NMR spectra (b) and Py-FTIR spectra(c), (d) of HZSM-5/Al2O3 catalyst recorded at 200 and 350 °C, respectively
With pyridine as a probe molecule, the distribution of acid sites over HZSM-5/Al2O3catalysts was investigated in detail by Py-FTIR.The results are displayed in Figure 3(c)-(d) and Table 2.Both calcined and steam-treated catalysts exhibit three characteristic bands at 1545, 1454, and 1490 cm-1.The first two bands can be respectively assigned to the adsorption of pyridine on Brønsted (B) and Lewis (L) acid sites,respectively, and the last one corresponds to the adsorption of pyridine on both B and L acid sites[18].The resulting concentrations of B and L acid sites,summarized in Table 2, show that the reduction of acidity after steam treatment was mainly caused by the loss of the dominant B acid sites.The concentration of B acid declined from 0.35 to 0.17 mmol/g, while the L acid increased slightly from 0.067 to 0.091 mmol/g.Similar trends are also observable for the medium &strong acid sites obtained from the Py-IR spectra after heating at 350 °C.Such an evolution of B and L acid sites suggests the transformation of the tetrahedral Al in the framework of HZSM-5, which is mainly responsible for the B acid sites[19], into extra-lattice alumina with steaming duration, fitting well with the increased (Si/Al)fratio after the steam treatment.

Table 2 Amount of Brønsted (B) and Lewis (L) acid sites determined by Py-FTIR on HZSM-5/Al2O3 catalysts
2.2 Cracking reactions
The product of FAMEs and methanol co-cracking over HZSM-5/Al2O3catalysts was divided into oil,aqueous, and gas phases after condensation.Under the conditions of 450 °C, 0.16 MPa and a space velocity of FAMEs at 1 h-1, the FAMEs conversion was 96.8% in the absence of methanol, and the selectivities of oil,aqueous, and gas phases were 60.3%, 7.5%, and 28.1%, respectively, as shown in Figure 4.Introducing 16% methanol improved the FAMEs conversion from 96.8% to 99.5%, suggesting the positive effect of methanol on the activity of HZSM-5/Al2O3towards FAMEs cracking.However, the conversion of FAMEs changed little with further increasing the methanol blending ratio, fluctuating around 99% as the methanol content was further increased to 100%.Meanwhile, oil and gas products decreased from 60.3% and 28.1% to 25.2% and 14.4%, respectively, with the rising methanol content.
Figure 5 further illustrates in detail the dependence of product distribution on the methanol blending ratios.Cracking FAMEs over the HZSM-5/Al2O3catalyst yielded aromatics as main products(54.7%), in which benzene, toluene, and xylene counted for 6.2%, 17.5%, and 19.2%, respectively.That is, the BTX selectivity added up to 42.9% for the cracking of FAMEs.Such a product distribution suggests that monocyclic aromatics were preferentially formed in the pores of HZSM-5 zeolite, despite the considerable amount of olefins (12.0%) and paraffins(33.3%).Different from the cracking of FAMEs,methanol cracking over HZSM-5/Al2O3catalysts yielded light paraffins as the main products.The selectivities of aromatics, olefins, and paraffins were 29.8%, 25.7%, and 44.5%, respectively.The lower aromatic selectivity and higher paraffin selectivity,compared with those for the cracking of FAMEs(54.7% and 33.3% in sequence), suggest a preferential transformation of olefins to paraffins over aromatics in the MTH.

Figure 4 Effect of the methanol blending ratios on the cocracking of FAMEs and CH3OH over HZSM-5/Al2O3 catalysts(reaction conditions: 450 °C, 0.16 MPa,WHSV (FAMEs) = 1 h-1)

Figure 5 Effect of the methanol blending ratios on the selectivities of (a) aromatics, (b) olefins, and (c) paraffins in the co-cracking of FAMEs and CH3OH over HZSM-5/Al2O3 catalysts (reaction conditions: 450 °C, 0.16 MPa, WHSV (FAMEs) = 1 h-1)
The different product distribution between the cracking of FAMEs and methanol originated probably in the mechanism involved in each reaction.Triglycerides and their derivatives, such as FAMEs,first undergo decomposition with the cleavage of C-O bonds in ester groups to produce long-chain hydrocarbons by releasing COxand H2O simultaneously[22], as schemed in Figure 6.The resulting hydrocarbons are then protonated to form carbenium ions at Brønsted acid sites of HZSM-5,followed by consecutive β-scissions and hydrogentransfer reactions to produce smaller hydrocarbon intermediates.Thereinto, the C6-9intermediates lead to either alkyl aromatics through cyclization and alkyl rearrangement or small molecules by the continuous cracking.The light C2-5olefins can oligomerize,cyclize, and aromatize to aromatics[5-7].Different from the cracking of FAMEs, the MTH proceeds via a dualcycle mechanism wherein the two cycles are dominated by olefins and aromatics, respectively[23-25].The olefinic cycle involves the methylation and cracking of olefins, and the aromatic cycle encompasses the methylation and dealkylation of aromatics.However,the two cycles are interrelated with each other through cyclization and hydrogen transfer reactions[26].That is,the cracking of both triglyceride and methanol can be subcategorized into elementary steps such as cracking and cyclization of olefins, hydrogen transfer,alkylation, and dealkylation of aromatics.It is,therefore, expected to regulate the product distribution by coupling FAMEs cracking and MTH processes since they require the same active sites and share similar reaction intermediates and products.

Figure 6 Proposed reaction pathway for the co-cracking of FAMEs and CH3OH over the HZSM-5/Al2O3 catalyst[5 - 7, 22 - 26]
The co-feeding of methanol changed, indeed, the product distribution of FAMEs cracking.The selectivity of aromatics decreased with increasing the methanol content from zero to 100%, but a reversed trend was observed for olefins.With an optimal methanol content of about 60%, the yields of desirable aromatics and olefins added up to 70.9%, much higher than cracking either reactant.Besides, the relative content of benzene in the aromatics dropped from 11.3% to 6.5% with the rising methanol content,suggesting the promoted methylation of benzene by the co-fed methanol.Different from aromatics and olefins,the selectivity of paraffins changed slightly with the blending ratios of methanol.The results indicated that the co-feeding of methanol degraded the formation of aromatics but promoted olefins in the cracking of FAMEs.According to Figure 6, aromatization processes provide hydrogen required for the cracking of FAMEs in the absence of methanol, which in turn produces aromatics as main products, as well as a considerable amount of coke[5-7].Co-feeding methanol with FAMEs, however, offers an efficient way to supply hydrogen and water.The surplus hydrogen released from the methanol cracking promotes the deep cracking of reaction intermediates and retards the aromatization of olefins, thereby resulting in the preferential formation of olefins in the co-cracking process.Moreover, the steam formed in methanol dehydration has also been reported to compete for the acid sites against hydrogen transfer reactions,attenuating the formation of aromatics and favoring the production of light olefins[27].
2.3 Attenuation of the deactivation
Besides the product distribution, the co-fed methanol also changed the deactivation behavior of the HZSM-5/Al2O3catalyst in the cracking of FAMEs, as shown in Figure 7.Despite the high selectivity of aromatics, the HZSM-5/Al2O3catalyst suffered a rapid deactivation in the cracking of FAMEs at a high space velocity of 4 h-1.The conversion of FAMEs increased initially from 93.8% to 96.4% in the first 4 h and declined finally to 39.8% after 12 h.By comparison,the methanol conversions in the cracking of methanol were almost constant during the first 12 h, remaining at around 97%.However, the initial conversions of FAMEs and methanol for the co-cracking reaction were 89.6% and 78.2%, respectively, much lower than cracking each reactant.The results suggest that the cracking of FAMEs and MTH may share and compete for the same active sites, namely, the Brønsted acid sites of HZSM-5 zeolite.Nevertheless, the co-cracking slowed down the catalyst deactivation rate.The FAMEs conversion decreased from 89.6% to 49.3%after 12 h but still higher than that for the cracking of FAMEs (39.8%).

Figure 7 Variation of FAMEs and CH3OH conversions with time-on-stream for the cracking of (a) FAMEs, (b) CH3OH, and (c) the mixture of FAMEs and CH3OH (conditions: 450 °C, 0.16 MPa, WHSV (FAMEs) = 4 h-1, WHSV (CH3OH) = 4 h-1)
Deactivation of HZSM-5 zeolites in the cracking reactions is generally related to the coke formation caused by the undesired deep dehydrogenation of aromatics due to the diffusion constraints imposed by the relatively small pores of HZSM-5 zeolites.The TG curves displayed in Figure 8 quantified the carbonaceous deposition on the used HZSM-5/Al2O3catalysts after reaction for 12 h.In the cracking of FAMEs, the total amount of deposited coke was 17.8%referred to the used catalyst weight.The weight loss of the TG curve, which occurred atca.300 °C and 510 °C with the respective mass drop of 5.8% and 12.0%,provides additional information about the nature of carbon deposition.The former loss was caused by the burn out of heavy hydrocarbons condensed on the external catalyst surface, whereas the latter corresponded to the combustion of polyaromatic coke[12].Compared with the cracking of FAMEs, the cofeeding of methanol with FAMEs reduced the carbonaceous deposition from 17.8% to 10.1%.In particular, the TG weight loss occurring above 400 °C dropped from 12.0% to 6.4%, suggesting much less polyaromatic coke deposited on the used catalyst for the co-cracking reaction.As discussed above, the water and hydrogen provided by MTH processes could promote the deep cracking of hydrocarbon intermediates, suppress the aromatization of olefins,and the following deep dehydrogenation to polyaromatics as coke precursors[28-30].Nevertheless,the co-cracking reaction still produced more coke than the MTH process (3.9%) under identical conditions,which might explain the continuous decrease in both FAMEs and methanol conversions with TOS in the cocracking reaction (Figure 7(c)).
2.4 Catalyst reactivation
The TG plots in Figure 8 confirmed the deposition of carbonaceous species on the used HZSM-5/Al2O3catalyst for the co-cracking of FAMEs and methanol.As a consequence, the surface area and pore volume of the spent catalyst decreased from 305 m2/g and 0.411 mL/g to 162 m2/g and 0.240 mL/g, respectively.However, the deactivated catalyst exhibited an identical XRD pattern to that of the fresh one, as seen in Figure 9(a), suggesting little change in the framework of HZSM-5 zeolites during the reaction.The spent HZSM-5/Al2O3catalyst was thus reactivated bycalcination at 600 °C, yielding comparable surface area and pore volume (332 m2/g and 0.423 mL/g) with the fresh one (345 m2/g and 0.434 mL/g), as shown in Figure 9(b) and listed in Table 1.The identical NH3-TPD spectra were also obtained between the fresh and reactivated catalysts (Figure 9(c)), indicating the restored acidity by coke combustion.Figure 9(d)presents the29Si NMR spectra of fresh, used, and reactivated HZSM-5/Al2O3composites.Compared with the fresh one, the used catalyst exhibited a broadened29Si NMR resonance peak, which was probably caused by the formation of coke in localized regions of zeolite and its distribution over the catalyst surface[31].Nevertheless, burning coke at 600 °C renewed the29Si NMR spectrum, suggesting the local environment of Si atoms was seldom affected by the coking-calcination cycle.Besides, the (Si/Al)fratios changed slightly from 31 to 32 (Table 1), implying a negligible dealumination of HZSM-5 zeolite in the co-cracking of FAMEs and methanol.

Figure 8 TG curves of the used HZSM-5/Al2O3 catalysts after reaction for 12 h with different feeds (reaction conditions:450 °C, 0.16 MPa, WHSV (FAMEs) = 4 h-1,WHSV (CH3OH) = 4 h-1)

Figure 9 XRD patterns (a), N2 adsorption-desorption isotherms (b), NH3-TPD profiles (c), and 29Si MAS NMR spectra(d) of the fresh, used, and reactivated HZSM-5/Al2O3 catalysts
The reactivated HZSM-5/Al2O3catalyst exhibited a comparable performance with the fresh one in the cocracking of FAMEs and methanol, as shown in Figure 10.The FAMEs conversion declined inevitably with reaction time from 89.6% and 86.6% to 49.3%and 47.8%, respectively, for the fresh and reactivated catalysts.A similar trend was also observed for the conversions of methanol on both catalysts.The restoration of catalyst activity to its original state and the simple regeneration process contribute to a promising application potential for the sustainable production of aromatics and olefins via the co-cracking of FAMEs and methanol over HZSM-5 zeolites.However, catalyst deactivation caused by coking remains still a challenge for the co-cracking reaction.Catalysts with long-term stability need to be further developed.

Figure 10 Reusability of the HZSM-5/Al2O3 catalyst for the co-cracking of FAMEs and methanol (conditions: 450 °C,0.16 MPa, WHSV (FAMEs) = 4 h-1, WHSV (CH3OH) = 4 h-1)
3 Conclusions
The effect of methanol on the cracking of FAMEs was probed over HZSM-5/Al2O3extrudates in terms of catalyst stability, selectivity, and reaction pathway.Catalytic cracking of FAMEs produced aromatics as main products, but the catalyst suffered a rapid deactivation due to coking.Co-feeding methanol with FAMEs enhanced the olefin selectivity greatly at the expense of aromatics, offering a practical method to regulate the product distribution by the co-cracking of FAMEs and methanol.Moreover, the co-fed methanol reduced coke markedly and attenuated the catalyst deactivation.The HZSM-5/Al2O3composite was still subject to deactivation in the co-cracking reaction, but the spent catalyst could be easily reactivated by calcination without noticeable degradation in catalytic performance.The findings may provide rationales for designing a feasible process to produce sustainable aromatics and olefins from triglycerides and their derivatives in the well-developed petroleum refineries.
