Co@NC选择性催化木糖氢解制备1,2-二元醇
2022-01-05李志坚刘琪英马隆龙
梁 缘 ,李志坚 ,刘琪英 ,马隆龙
(1.中国科学技术大学工程科学学院,安徽 合肥 230026;2.中国科学院广州能源研究所,广东 广州 510640;3.中国科学技术大学纳米学院,江苏 苏州 215123;4.中国科学院可再生能源重点实验室,广东 广州 510640)
在传统化石能源日渐匮乏以及过度使用化石燃料引发的环境问题的压力下,人们一直在不懈地寻找低成本的可再生能源[1]。木质纤维素生物质是地球上储量最大的碳质可再生资源,主要由纤维素(40%-50%)、半纤维素(23%-32%)和木质素(15%-25%)构成[2]。其中木糖作为半纤维素的主要成分,富含羟基官能团,是制备平台化学品的理想原料,其开发和利用引起了社会各界的广泛关注。
1,2-戊二醇(1,2-PeD)是一种重要的化工中间体,年消耗量超过2000吨,其中,80%转化为高效低毒、广泛使用的杀菌剂丙环唑,其余部分用于化妆品合成或医药应用[3]。目前,1,2-PeD的工业生产主要包括1-戊烯氧化成1,2-环氧戊烯及后续水解。但是该过程使用大量的强氧化剂如次氯酸钠和过氧乙酸,除了对环境产生污染外,也会对反应设备产生腐蚀。使用可再生的木糖直接加氢脱氧制备1,2-PeD是很有前景的研究策略,但C-C键在高温条件下易断裂,使得生成长链1,2-二元醇具有挑战性。Ordomsky等[4]使用Ru/C协同Amberlyst-15催化剂,催化木糖脱水、加氢得到1,2-PeD(产率10%)。Wang等[5]使用Ru/C协同Nb2O5固体酸催化剂,在水/环己烷/γ-戊内酯体系中对1,2-PeD的最佳收率为19.1%。然而,上述木糖制备1,2-PeD的研究中应用了贵金属催化剂,考虑到贵金属高昂的使用成本和有限的储量,亟待开发出非贵金属催化剂以实现木糖向1,2-PeD的直接转化。
乙二醇(EG)和 1,2-丙二醇(1,2-PG)是重要的化工原料,可用于生产聚酯纤维和树脂材料,亦广泛应用于医药、食品等领域。目前,主要由石油衍生产品乙烯、丙烯和他们的环氧中间体生产EG和1,2-PG。应用木糖及其衍生物制备EG和1,2-PG符合可持续性和绿色化学的理念。Sun等[6]使用Ru/C催化剂,在Ca(OH)2的存在下水相催化木糖醇,EG和1,2-PG的总收率达到57.3%。碱的存在促进氢解反应[7],得到中间产物乙醇醛和甘油醛,后续发生加氢反应得到上述两种二元醇。但贵金属催化剂由于高昂的成本限制了其大规模应用,并且Ca(OH)2的添加对设备的耐腐蚀性提出了更高的要求。
将过渡金属包裹于保护性碳壳层是一种具有前景的催化剂设计手段,可适应生物质催化炼制苛刻的反应环境。表面碳层避免内部金属与外界直接接触,使催化剂在强酸、高温等苛刻条件下具有较好的稳定性;同时通过石墨良好的电子传输能力,可调控内部金属的催化活性[8,9]。这种策略目前已在电催化、光催化等领域证明了其可行性[10-13]。此外,在碳骨架中掺杂杂原子如N、P、S等可以调节石墨的共轭电子结构,引入表面缺陷位和丰富反应位点,可进一步调控催化剂的反应性能[14-16]。
本研究设计制备了N掺杂碳纳米管包裹过渡金属Co@NC催化剂,在水相中选择性催化木糖氢解制备二元醇(EG、1,2-PG和1,2-PeD),总产率达70%,其中,对1,2-戊二醇的收率27.4%,为文献报道最好结果。木糖通过Retro-aldol反应得到乙醇醛和丙酮醇,随后对不饱和C=O键加氢得到乙二醇和1,2-丙二醇;另一方面,通过保留木糖的碳链结构,选择性断裂C-O键,得到1,2-戊二醇。对比了不同焙烧温度下Co@NC催化剂的表征结果和催化活性,考察了反应温度、反应时间、氢气压力对产率的影响,探讨了可能的反应路径和协同催化作用机制。
1 实验部分
1.1 原料和试剂
催化剂合成所用Co(NO3)2·6H2O,Ni(NO3)2·6H2O,三聚氰胺和活性炭等采购于阿拉丁生化科技有限公司,D-木糖、D-木酮糖、木糖醇、糠醛、糠醇、乙二醇、1,2-丙二醇、1,2-戊二醇、乙醇醛、丙酮醇、丙酮醛等采购于麦克林生化科技有限公司。所有试剂均为AR级,未经进一步纯化直接使用。
1.2 催化剂的制备
本文涉及的催化剂主要包括包裹型催化剂M@NC 和M@C(其中,M= Co、Ni、Fe),负载型催化剂Co/C,无金属催化剂N-活性炭。
使用bottom-up法制备包裹型催化剂。第一步为前驱体制备,对于M@NC催化剂,将0.03 mol的M金属的硝酸盐和0.09 mol的三聚氰胺 (C3N6H6)溶于50 mL去离子水中,强烈搅拌形成均匀溶液。将该混合物在70 ℃下搅拌至可见水全部蒸发,转移到80 ℃鼓风干燥箱干燥24 h。所得固体充分研磨后,在 N2氛围,500-800 ℃ 下焙烧 3 h,N2流量为30 mL/min,加热速率5 ℃/min。所得催化剂标记为M@NC-t,M为所用金属,t为煅烧温度。
不掺 N 的M@C(M= Fe、Co、Ni)催化剂制备方法如下:0.03 mol的M金属硝酸盐和0.09 mol柠檬酸(C6H8O7)溶于20 mL去离子水中,在300 r/min的搅拌速率下形成均匀的溶液。将该溶液在70 ℃下搅拌蒸干至胶状,转移烧杯至80 ℃鼓风干燥箱中干燥24 h。所得固体在研钵中充分研磨,在N2气氛下以5 ℃/min的升温速率升至600 ℃,焙烧3 h。
使用浸渍法制备负载型Co/C催化剂。载体活性炭在搅拌下加入至1 mol/L的硝酸钴水溶液中,室温搅拌24 h,再放在100 ℃鼓风干燥箱中干燥12 h。所得黑色固体在H2气氛下于450 ℃还原 3 h,H2流量 30 mL/min,升温速率 5 ℃/min。
无金属催化剂N-活性炭(N-AC)的制备方法如下:将4 g三聚氰胺溶于30 mL水中,在70 ℃下加热0.5 h至三聚氰胺溶解,得到乳白色溶液。称量4 g活性炭加入烧杯,在该温度下加热12 h,然后将烧杯转移至80 ℃烘箱中干燥24 h。所得黑色固体在30 mL/min的N2气流中加热至600 ℃保持 3 h,升温速率 5 ℃/min。
1.3 催化剂的表征
催化剂的晶型及物相结构由XRD测定得到。仪器型号为荷兰X Pert Pro MPD(PW3040/60),采用 Cu靶和Kα辐射源(λ= 0.154 nm),管电压(靶压)和管电流分别为40 kV和40 mA,扫描角度5°-80°。催化剂表面的价态组成、元素分布由XPS测定。所使用的仪器型号为美国Thermo Fisher Scientific Escalab 250 XI光电子能谱仪,射线源为 MgKα(1235.6 eV, 电压 10 kV, 电流 23 mA)。使用284.8 eV的C 1s线校正元素结合能,对荷电效应引起的位移进行校正。催化剂的形貌特征由TEM测定。所使用的仪器型号为日本电子株式会所的JEM-2100F场发射高分辨透射电镜,操作电压为200 kV。催化剂的BET面积通过在-196 ℃下的N2吸附-解吸等温线测定,并使用Barrett-Joyner-Halenda (BJH) 方法评估孔径分布。使用FT-IR测定催化剂表面的官能团信息,仪器型号为Nicolet iS50,400-4000 cm-1扫描。催化剂的石墨化程度和无序程度由Raman测定得到。仪器型号为LabRAM HR800-LS55型激光共聚焦拉曼光谱仪,激光波长为525 nm,500-3000 cm-1扫描。反应过程中的金属流失使用ICP-AES测定,仪器型号为Perkin-Elmer OPTIMA 8000。催化剂表面的碱性和酸性位数量分别用CO2-TPD和NH3-TPD测定得到,仪器为天津Xianquan公司的TP-5080。样品在500 ℃,He气氛下预处理1 h以除去表面不稳定物质,冷却至50 ℃吸附CO2或NH3至饱和,再次切换为He气氛,以10 ℃/min的升温速率升温至800 ℃,记录出口处TCD信号。CO2或NH3的脱附量以250 μL的定量环定量。
1.4 催化性能评估及稳定性考察
木糖及可能反应中间体的氢解实验均在50 mL不锈钢高压釜(MS-50,安徽科幂机械科技有限公司,安徽合肥)中进行。典型的实验过程如下:0.2 g木糖、0.07 g催化剂和20 mL去离子水置于高压反应釜。密封反应釜,用氢气置换四次以除去釜内残余的空气,在室温下将氢气压力升至3 MPa。开启加热,以10 ℃/min的速率升温至200 ℃,保持3 h,搅拌转速为800 r/min。反应结束后,用冰水混合物将反应釜迅速冷却至室温,通过过滤、离心,收集液体产物和固体部分。
为测试催化剂的循环使用性能,每次反应后将固体部分用乙醇、去离子水洗至中性,在-37 ℃真空冷冻干燥12 h,直接用于下一次反应。
1.5 反应产物分析
分析前液相产物由0.22 μm水相滤膜过滤。液相产物的定性分析主要采用气相色谱-质谱联用仪(GC-MS,Thermal Trace 1300ISQ QD,INNOWAX色谱柱 30 m × 0.32 mm × 1.0 μm 和 Polaris Q 离子色谱)进行分析。气相色谱检测器温度为260 ℃,He载气流量40 mL/min;质谱仪传输通道和离子源温度分别为260和240 ℃。质谱仪扫描电压为70 eV,33-400 amu扫描,扫描 0.2 s。根据 GCMS给出的推测分子式,在GC测试中检验样品的标准溶液,与反应液中的出峰时间进行对比,验证、确定产物结构。
液相产物中的醇类产物的定量分析主要通过气相色谱仪(GC,Shimadzu 2014C)完成,使用INNOWAX 色谱柱(30 m × 0.32 mm,1.0 μm),FID检测器。分析过程中的程序升温设定为:在起始温度60 ℃下维持2 min,随后以10 ℃/min的升温速率升至260 ℃,在终温下保持10 min。液相产物中木糖、木糖醇等的定量分析主要通过高效液相色谱仪(HPLC,Waters 2695,Shodex Sugar SH1011色谱柱,示差检测器),流动相为5 mmol/L的硫酸溶液,流量0.5 mL/min,柱温50 ℃,采用外标法进行定量。
原料转化率Conversion (%)和产物收率Yield(C-mol%)的计算方法如下:
木糖的转化率:

式中,Bxylose初始加入的木糖质量,Axylose表示反应后木糖的剩余质量。
木糖反应各种产物的收率基于碳原子数计算:

式中,ni表示第i种产物的物质的量,ki表示第i种产物所含碳原子数,MC5H10O5表示木糖的相对分子质量,mxylose表示加入的木糖质量。
2 结果和讨论
2.1 催化剂的表征
不同温度下焙烧的Co@NC催化剂的XRD谱图如图1所示。除了Co@NC-500,其他温度下制备的样品都显示了在2θ= 26.5°的炭特征衍射峰。对于600-800 ℃下制备的Co@NC,可以观察到2θ= 44.2°、51.5°和 73.9°的 Co 特征峰,分别对应立方相 Co的 (111)、(002) 和 (022)的晶面衍射峰(JCPDS 96-901-0969)[17]。在更高的煅烧温度(800 ℃)下,Co的特征衍射峰强度增加,这可能是因为在高温下碳纳米管破裂,Co颗粒发生了团聚现象,粒径增大,导致相应的峰强度增加。

图1 不同温度下煅烧的Co@NC催化剂的XRD谱图Figure 1 XRD patterns of Co@NC prepared at different calcination temperature
不同制备温度Co@NC催化剂的TEM数据如图2所示。对于Co@NC-500,500 ℃的焙烧温度不足以使碳氮中间体以Co为成核点进行卷曲以形成碳纳米管结构,因此,仅得到一些片状的石墨炭层。当焙烧温度升至600 ℃及以上时,有机物分解、炭化,在金属颗粒的作用下发生卷曲,形成碳纳米管包裹金属结构。当焙烧温度达到800 ℃时,碳纳米管直径增大,纳米管分布更稀疏,Co粒子发生团聚,粒径增大[14]。

图2 不同焙烧温度Co@NC催化剂的TEM照片Figure 2 TEM images of Co@NC synthesized at (a), (b) 500 ℃, (c), (d) 600 ℃, (e), (f) 700 ℃ and (g), (h) 800 ℃
通过XPS研究了催化剂表面的元素和价态组成。如图3所示,Co@NC催化剂表面的N含量随焙烧温度的升高而下降,其原因是高温下更多的N物种分解为NO、NH3等逸出[18]。图4(a)是不同催化剂的N 1s谱图,其中,N的种类包括N-Co和吡啶N(398.4 eV)、石墨N(401.2 eV)、氧化N(404.1 eV)[19]。N-Co的峰与吡啶N的峰相近,因此,很难被进一步区分[20]。从图中可以看出,随着焙烧温度的升高,N-Co/吡啶N的比例下降,石墨N的比例随着温度升高而增大,说明在高温下会发生其他N物种向石墨N的转化,这是因为相比于其他类型的N,石墨N具有更好的热稳定性[15]。

图3 不同温度焙烧的Co@NC催化剂的XPS谱图Figure 3 XPS spectra of Co@NC prepared at different temperature
不同温度焙烧的催化剂的Co 2p的XPS能谱图如图4 (b)所示,其中,位于778.8、780.2、782.7 eV的特征峰分别属于Co0、CoII和CoIII[21]。在热解过程中随着焙烧温度的提高,碳物种会转变为还原性气体如CO、CH4等逸出,这些气体可以作为还原剂对金属进行还原,将CoII和CoIII还原为Co0[22]。该零价Co可以在空气环境中得以保留,说明包裹炭层可以防止内部金属被氧化[23]。

图4 不同焙烧温度Co@NC催化剂的(a) N 1s 能谱和(b) Co 2p 能谱图Figure 4 (a) N 1 s and (b) Co 2 p spectra of Co@NC synthesized at different temperature
不同焙烧温度制备的Co@NC催化剂的氮气吸附-脱附曲线和孔径分布如图5所示。500 ℃制备的催化剂中未检测到孔的存在,没有观测到对氮气的明显的吸附-脱附现象。在600-800 ℃下制备的催化剂显示了明显的IV型吸附-脱附曲线[14]。BET比表面积和孔的参数如表1所示。随着焙烧温度由600 ℃升高至700 ℃,BET比表面积和孔体积增加;进一步升高焙烧温度,孔体积继续增大,但催化剂BET比表面积变化不明显。孔容增加可能是因为更高温度下石墨结构的破裂,形成孔体积更大的多孔结构[24]。

表1 Co@NC催化剂的BET数据和孔结构参数Table 1 BET surface area and pores of Co@NC catalysts

图5 不同焙烧温度Co@NC催化剂的(a)氮气吸附-脱附曲线和(b)孔径分布Figure 5 (a) The nitrogen adsorption-desorption isotherms and (b) the pore size distribution of Co@NC catalysts calcinated at different temperature
催化剂的FT-IR谱图如图6所示。其中,3000到3650 cm-1出现的峰为N-H键和表面吸附的H2O的伸缩振动[13],在1150-1650 cm-1的峰为C-N杂环振动[25],说明N在C骨架中的配位。使用CO2-TPD和NH3-TPD对催化剂表面的碱性/酸性位数量进行表征,结果如表2所示。对于CO2-TPD,催化剂的碱性位点包括在150 ℃的弱碱位点和580 ℃的强碱位点。随着焙烧温度的升高,Co@NC的碱性位点数量下降,这是因为催化剂的碱性位点主要来源于N的掺杂,焙烧温度升高导致N以气态物种逸出,使得催化剂的总碱量减少。催化剂表面的酸性位数量使用NH3-TPD进行表征。根据相关文献,催化剂表面的酸性可能来自于Coδ+提供的酸性位点[26]。

表2 Co@NC催化剂的表面酸/碱量Table 2 Surface acid/base amounts of Co@NC catalysts

图6 不同焙烧温度Co@NC的FT-IR谱图Figure 6 FT-IR spectra of Co@NC catalysts synthesized at different temperature
使用TG-MS对不同温度焙烧的Co@NC进行分析。从图7中可以看出,Co@NC-500在100 ℃附近和600-700 ℃出现了约10%和55%的质量损失,而600-800 ℃焙烧的Co@NC质量损失较少,说明在500-600 ℃的焙烧过程中大量基团分解逸出。出口气体主要包括CH4(m/z= 16)、H2O(m/z= 18)、 N2/CO (m/z= 28)、 NO (m/z= 30)和N2O/CO2(m/z= 44)。对比图7和图8发现,在100 ℃左右的质量损失主要为H2O,在700 ℃左右的质量损失主要为N2/CO2。结合催化剂的CO2-TPD数据认为,在焙烧温度由500 ℃升至600 ℃的过程中,大量N物种以NH3和N2形式逸出,导致N含量及碱量的显著降低。

图7 不同焙烧温度Co@NC催化剂的TG-MS谱图Figure 7 TG-MS analysis of Co@NC synthesized at different temperature

图8 Co@NC分解过程中出口气体的检测Figure 8 Outlet gases from the decomposition of Co@NC
通过Raman研究了不同催化剂的石墨化程度。如图9所示,石墨烯的D峰位于1345 cm-1附近,代表石墨烯结构中的结构和边缘缺陷。石墨烯的G峰位于1575 cm-1附近,是由一阶E2g声子平面振动引起,反映炭材料的对称性和有序度[16]。石墨烯的2D峰位于2675 cm-1,属于双声子共振拉曼峰。随着焙烧温度的升高,D峰与G峰的强度比下降。更高的焙烧温度减少了N的掺杂量,减少了表面缺陷位点,使得D峰强度下降;而另一方面,在更高的焙烧温度下,会得到更有序的、热力学更稳定的石墨烯炭,使G峰信号增强[27]。所以焙烧温度升高,ID/IG值下降。

图9 不同焙烧温度Co@NC催化剂的Raman谱图Figure 9 Raman spectra of Co@NC calcinated at different temperature
2.2 Co@NC催化木糖氢解制备1,2–二元醇
Co@NC催化剂水相转化木糖的转化率和液相产物收率如表3所示。Co@NC-500催化剂对木糖转化率仅37.3%,这可能是因为Co@NC-500的BET比表面积仅为3.4 m2/g,而600-800 ℃焙烧的Co@NC的BET比表面积为194-275.3 m2/g。因此,Co@NC-500较低的BET比表面积限制了异相催化过程,导致其较低的木糖转化率。此外,在Co@NC-500反应后的溶液中观察到了结焦,这可能是因为其最高的碱性位数量促进糖类物质发生聚合反应[28]。在Co@NC-600催化剂上获得了最佳的催化效果,木糖的转化率提高到了95%以上,EG、1,2-PG和1,2-戊二醇的产率分别达到了17.6%、25.1%和27.4%,总二元醇产率达到70.1%。此外有少量的环戊酮、丙酮醇和四氢糠醇作为副产物出现。对于更高温度下制备的Co@NC,二元醇的总收率下降,但丙酮醇产率升高,且对于600-800 ℃下制备的Co@NC,其1,2-PG+丙酮醇的总收率相近,分别为28%、29.2%和29.6%。丙酮醇可以通过加氢反应得到1,2-PG,因此,更高温度焙烧的Co@NC催化剂对二元醇收率的下降可能是因为其加氢能力的减弱,抑制了酮醇加氢生成二元醇。

表3 不同N掺杂碳包裹型催化剂在木糖氢解反应中的催化性能Table 3 Xylose conversion to 1,2-diols over different N-doped carbon encapsulated metal catalysts
对比了不同金属催化剂对木糖的催化效果。在Ni@NC催化剂上,木糖的转化率为82.4%,二元醇收率较低,而环戊酮、丙酮醇和四氢糠醇的收率分别达到15.0%、20.5%和10.3%。环戊酮和四氢糠醇被认为是糠醛的下游产品,其中,环戊酮是糠醛通过Piancatelli重排得到[29],而四氢糠醇是糠醛完全加氢的产物[30]。说明Ni@NC催化剂可以催化木糖脱水得到糠醛,并且促进糠醛向环戊酮和四氢糠醇的转化。Fe@NC对木糖的催化活性较低,且仅得到10.5%的丙酮醇。
实验制备了不掺N的Co@C、Ni@C和Fe@C催化剂,以及无金属的N-活性炭催化剂,进行木糖氢解实验以探究其催化活性,结果在表4中显示。相比于M@NC,M@C的加氢活性更高,以木糖为原料时得到了高产率的完全加氢产物木糖醇。对于N-活性炭,得到了13.8%的环戊酮和20.4%的丙酮醇。将活性炭与三聚氰胺共热解后,相当于在活性炭表面负载了一部分的N物种,给催化剂提供了一定的碱性活性位点,一定程度上促进Retro-aldol反应得到C-C键裂解产物丙酮醇[31],但因没有金属的存在,N-活性炭的加氢活性较弱,没有木糖醇或二元醇的生成。

表4 不同催化剂上木糖的氢解性能Table 4 Xylose hydrogenolysis catalyzed by different catalysts
对于Co@NC-600催化剂,考察了不同反应条件对1,2-二元醇产率的影响,如图10所示。研究发现,在更高的反应温度下,1,2-PeD的产率显著降低,这可能是因为在C-C键在高温下发生的裂解。延长反应时间,同样观测到了1,2-PeD产率的下降,原因是长链二元醇相比乙二醇和丙二醇等小分子醇更不稳定,在较长的反应时间下分解为其他物种[32]。图10(c)反映了H2压力的影响,氢气压力较低时,H2活化解离的H减少,加氢反应被抑制,二元醇类产率下降。在最佳反应条件下,1,2-PeD的产率高于文献报道的非贵金属的最佳产率,如表5所示。

表5 1,2-PeD收率与文献对比Table 5 Comparison of 1,2-PeD yield in this work with reported references

图10 Co@NC-600催化剂上不同反应条件下木糖氢解反应性能Figure 10 Effect of (a) reaction temperature, (b) reaction time and (c) H2 pressure on the selective hydrogenolysis of xylose over Co@NC-600
2.3 催化剂循环使用性能
通过进行五次循环实验考察了Co@NC的循环使用性能,在每次循环后离心收集固体催化剂,经水洗、乙醇洗后冷冻干燥后直接用于下一次反应。反应结果如图11所示,从图中可以看出去,木糖的转化率和二元醇产率未明显变化,说明在五次循环反应中,Co@NC催化剂具有优异的稳定性。

图11 Co@NC催化木糖转化的循环使用性能Figure 11 Cycle test of Co@NC for xylose conversion.
通过TEM研究了催化剂在反应前后的形貌特征,结果如图12所示。发现使用前后的Co@NC催化剂的仍保持碳纳米管形貌,金属未发生团簇,是其具有优异水热稳定性的原因。对五次使用后液相的ICP-AES进行分析,液体中Co的流失可忽略不计(< 1.0×10-7mg/L),说明碳纳米管的包裹可以有效防止高温高压和水相反应环境下Co的浸出。

图12 反应前后Co@NC催化剂的TEM照片Figure 12 TEM results of (a), (b) fresh Co@NC-600, (c), (d) spent Co@NC-600 after five runs
2.4 Co@NC催化木糖氢解制备二元醇的反应路径与催化机理
为了探究Co@NC催化剂催化木糖转化为EG、1,2-PG和1,2-PeD的反应途径,使用Co@NC催化可能的反应中间体如木糖醇、丙酮醇、糠醇等,其结果如表6所示。相比于含不饱和C=O键的木糖,木糖醇的稳定性显著提高,在该水热条件下仅有25.2%的转化率,未观察到对醇类产物明显提升的选择性。木酮糖作底物时主要得到EG和1,2-PG。选用乙醇醛做原料时,得到了近100%的转化率和93.7%的EG选择性,说明乙醇醛是EG的前驱体,在该催化体系下,乙醇醛可以在Co@NC的催化下加氢得到二元醇。用丙酮醇和丙酮醛验证1,2-PG的来源。丙酮醇的转化率和对1,2-PG的产率分别达到了98.2%和95.3%,说明丙酮醇的羰基可加氢得到1,2-PG;丙酮醛为原料时获得95.6%的转化率,但对1,2-PG的选择性仅有39.1%,此外得到了19.3%产率的丙酮醇。对于丙酮醛的加氢,受到空间位阻和醛、酮活性差异的影响,首先对醛基进行亲核加成,得到丙酮醇后才可以进行羰基的加氢,相比于直接采用丙酮醇作原料,两步连续反应以及醛类在高温下可能发生的副反应降低了对1,2-PG的选择性。以糠醛和糠醇为原料时,通过Piancatelli重排反应得到了高产率的环戊酮和环戊醇。此外实验使用六碳糖对催化剂的活性进行探究,发现使用果糖原料时得到了丙酮醇和1,2-PG,产率分别为30.1%和10.4%,说明该催化体系可以促进酮糖发生Retro-aldol反应,可进一步解释乙醇醛和丙酮醇的生成。

表6 不同原料下的产物分布Table 6 Product distribution using various possible intermediates
为探究木糖和木酮糖之间的异构化反应,在N2氛围下使用Co@NC-600以木糖为原料进行反应,如图13所示。碱性位点可以促进糖类的异构[33],在较短的反应时间的反应液内检测到了一定量的木酮糖,说明Co@NC催化剂可以促进木糖向木酮糖的异构。

图13 短反应时间下木糖向木酮糖的异构Figure 13 Isomerization of xylose to xylulose at short reaction time
基于上述讨论的结果,得到了如机理图14所示的反应路径。在Co@NC催化剂的碱性位点作用下,木糖发生了醛酮异构得到了木酮糖,木酮糖随后发生Retro-aldol反应,C-C键断裂得到乙醇醛和丙酮醇。虽然有石墨的包裹,但通过碳壳层良好的电子传输效应可以实现内部金属Co的加氢能力,得到乙二醇和1,2-丙二醇。在催化剂的酸性位点和金属加氢位点的作用下,木糖发生选择性加氢脱氧反应(HDO)得到1,2-戊二醇。此外,木糖脱水得到糠醛,通过加氢、重排等反应得到四氢糠醇和环戊酮等副产物。在该催化过程中存在酸、碱和金属的协同催化:Coδ+提供的酸性位点一方面促进木糖脱水得到糠醛,经由后续加氢、重排等反应得到四氢糠醇、环戊酮等;另一方面与金属Co加氢位点协同催化木糖的加氢脱氧反应,得到1,2-戊二醇;N掺杂提供的碱性位点促进木糖向木酮糖的异构,木酮糖经Retro-aldol反应得到乙醇醛和丙酮醇,通过金属Co加氢得到乙二醇和1,2-丙二醇。
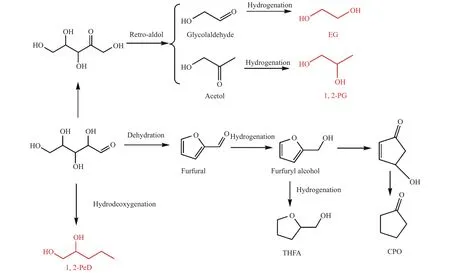
图14 木糖催化氢解的可能反应途径示意图Figure 14 Possible reaction route for xylose hydrogenolysis to vicinal diols
3 结 论
制备了具有加氢和异构活性的Co@NC催化剂,在水相中催化木糖氢解制备1,2-二元醇类产物。焙烧温度对Co@NC催化剂的结构组成和催化效果有显著影响。在Co@NC-600催化剂上获得了最高的催化活性,邻二醇总收率70.1%,其中,1,2-戊二醇的收率高于文献报道的非贵金属收率(27.4% 和 19.1%)。N的掺杂为Co@NC提供了碱性位点,碱促进木糖向木酮糖的异构,随后发生Retro-aldol反应生成乙醇醛和丙酮醇,最后发生加氢反应得到产物乙二醇和1,2-丙二醇;该工作实现了长链邻二醇的生成,木糖经由加氢脱氧反应生成1,2-戊二醇。该类包裹型非贵金属催化剂具有优异的水热稳定性。本研究工作为生物基1,2-二元醇,尤其是长链二元醇产物的高效制备提供了新思路。
