核-壳型PEM电解水制氢阳极铱基催化剂的研究进展
2021-12-22苏倩倩米万良顾方伟郑路凡赵熙康
苏倩倩,米万良,张 聪,顾方伟,郑路凡,赵熙康,魏 强
(1.中国石化 石油化工科学研究院,北京 100083;2.中国石油大学(北京)化学工程与环境学院,北京 102249)
氢气是目前最具前景的储能介质,作为一种理想的能源载体它具有燃烧热量高[1]、燃烧无污染[2-3]、来源广[4]、可再生等优点。目前已经有多种氢气制备方法[5],可由各种可再生或不可再生能源转化制得,其中通过化石燃料制氢过程中产生大量二氧化碳,制得的氢气多为灰氢[6],通过可再生能源制氢过程中不产生污染物,制得的氢气多为绿氢或蓝氢。根据中国氢能发展要求,绿氢将是中国制氢发展的方向[7]。基于可再生能源电力的电解水制氢规模大、效率高、无污染、产氢纯度高,是生产绿氢的可靠技术。
基于可再生能源电力的质子交换膜(PEM)电解水制氢成本较高,目前尚不具备大规模发展的优势,亟待寻找降低电解水制氢成本的方法。制约PEM电解水发展的因素主要可以分为3个方面:一方面是质子交换膜成本较高[8],目前常用的全氟磺酸质子交换膜多为杜邦公司的Nafion®[9]、Fumapem®等类固体聚合物膜,价格昂贵,需要进一步改进制备工艺、研究低成本的质子交换膜;第二方面是集电器和双极板成本高[10];第三方面是制备膜电极所用的阴阳极催化剂成本高,满足阳极苛刻反应条件的催化剂主要局限于贵金属衍生物[11-12]。
阳极催化剂成本居高不下的根源在于阳极电极反应的过电势较高,同时处于氧化和酸性环境下,对催化材料的要求较苛刻。阳极析氧反应(OER)是一个四电子的电极反应,是PEM水电解反应的速率控制步骤,四电子转移的缓慢动力学产生了较高的析氧反应过电位,高过电位和严酷的酸性环境增加了对催化剂化学稳定性和催化活性的要求[13]。在众多电极材料中Ir基催化剂脱颖而出,凭借其较高稳定性、低过电位的优势成为电解水阳极反应中最常用的贵金属催化剂。在OER过程中Ir的催化作用机理如图1所示[14-16],从Ir(OH)3开始,通过发生一步去质子化步骤使Ir(OH)3转变为IrO(OH)2,再经过两步连续的去质子化步骤之后得到IrO3,此时Ir处于六价状态,接下来有2种途径:一种途径是吸收水分子,释放氧气,自身被还原成三价Ir回到循环的起始位置;另一种途径是吸收水分子,以IrO4-的形式溶解在电解液中,这一种途径是不被期望发生的,这意味着催化剂的稳定性遭到破坏。
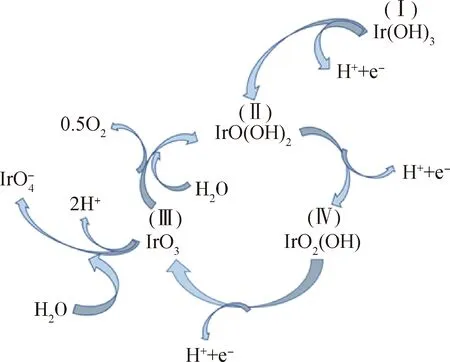
图1 铱电极上电荷存储和氧析出模型[14-16]Fig.1 Charge storage on iridium electrode and oxygen evolution model[14-16]
由于Ir资源稀少、价格昂贵,因此开发出提高Ir利用率的高活性催化剂是重中之重[17]。为了降低Ir的用量,提高催化剂的活性和稳定性,研究者们通过开发掺杂型[18-20]、负载型[21-22]等Ir基催化剂来提高Ir的利用率,改进催化剂的性能,进而降低催化剂的成本。Cheng等[23]通过利用Adams法将Ir和Ru掺杂制备出了IrxRu1-xO2(x=0.2、0.4、0.6)复合催化剂,结果表明,Ir与Ru的掺杂提高了Ru的稳定性,其中Ir0.2Ru0.8O2的析氧反应活性最佳,在电流密度为1 A/cm2时电压为1.622 V。Li等[24]利用添加表面活性剂制备出了双金属氧化物IrxSn1-xO2(x=1、0.67、0.52),形成无定形结构的固溶体,分散性良好,研究结果表明,Ir0.52Sn0.48O2的OER活性最高,在电流密度为1 A/cm2时电池电压为1.631 V。Xu等[25]通过将IrRu负载在Te上改变了IrRu的电子结构,使d带中心提升,OER催化活性和稳定性均有所提高。Li等[26]通过将Ir负载在TiN上大大降低了贵金属的用量,并且TiN具有抑制Ir团聚和溶解的作用,以及通过向Ir转移电子使得Ir的d带中心下移,解决了氧物种在金属上吸附能过大的问题,增加了Ir/TiN催化剂的稳定性和催化活性。综上所述,通过将Ir与其他金属复合或者引入高比表面积、高稳定性的载体可以降低Ir的用量,提高催化剂的活性。
研究者们也把目标转向了非贵金属催化剂[27-28],但同样都面临着在催化剂的使用过程中伴随着金属溶解的问题,或者缓慢动力学限制了非贵金属催化剂的实际应用。因此如何开发出高性能、低载量、高稳定性的电催化剂,以减少对贵金属的依赖,降低PEM电解水制氢的成本,已成为PEM电解水制氢研究领域内亟需解决的难题。
从1990年以来,广泛应用于光电化学、生物传感器和催化等领域的纳米核-壳结构催化剂[29-30]的制备成为了研究热点。在PEM电解水制氢领域降低贵金属用量和提高催化剂活性及稳定性的研究中,核-壳结构型催化剂发挥了重要作用。核-壳结构催化剂是由至少两种或两种以上的物质组成,一种物质形成核,另一种物质包裹在外形成壳,一般记作“核@壳”[31],得益于其表面应变和原子附近影响核-壳之间的电荷转移,使得这种结构具有独特的物理化学性质,提高了在OER过程中的稳定性和催化活性。电催化过程是一个表面反应,只有分布在催化剂表面的活性位点才能够参与反应[32],可以充分利用核-壳结构将贵金属包裹在外形成壳,内核用其他非贵金属物质来代替,最大限度地增加了贵金属与反应物的接触,同时又减少了贵金属的使用量,并通过核-壳协同作用(如调节电子效应和表面应变效应)增强了催化剂的活性和稳定性[33]。
1 PEM电解水制氢原理
PEM电解水制氢采用固体聚合物质子交换膜作为隔膜,替换了传统碱性电解水通常使用的石棉隔膜和液体电解质,提高了电解水制氢的安全性及电解效率[34]。PEM电解水阴阳两极发生的电化学反应见式(1)~(3)。
总反应:
2H2O→2H2+O2
(1)
阴极:
2H++2e-→H2
(2)
阳极:
2H2O→O2+4H++4e-
(3)
PEM水电解制氢工作原理如图2所示[35],阳极附近的水分子在电压的作用下失去电子发生氧化析氧反应(OER),电子通过外电路转移到阴极,氢质子通过质子交换膜迁移到阴极得电子发生还原析氢反应(HER)。HER是一个简单的半反应,涉及到的中间体较少,反应动力学相对于OER而言较快。OER是一个涉及4电子的过程,反应中间体较多,反应动力学缓慢,产生的过电势较高,是电解水过程中过电势的主要来源,成为电解水过程的速率控制步骤。OER反应过程复杂,所以动力学机理的研究不如HER透彻,目前比较被认同的OER反应机理如式(4)~(9)所示[36]。
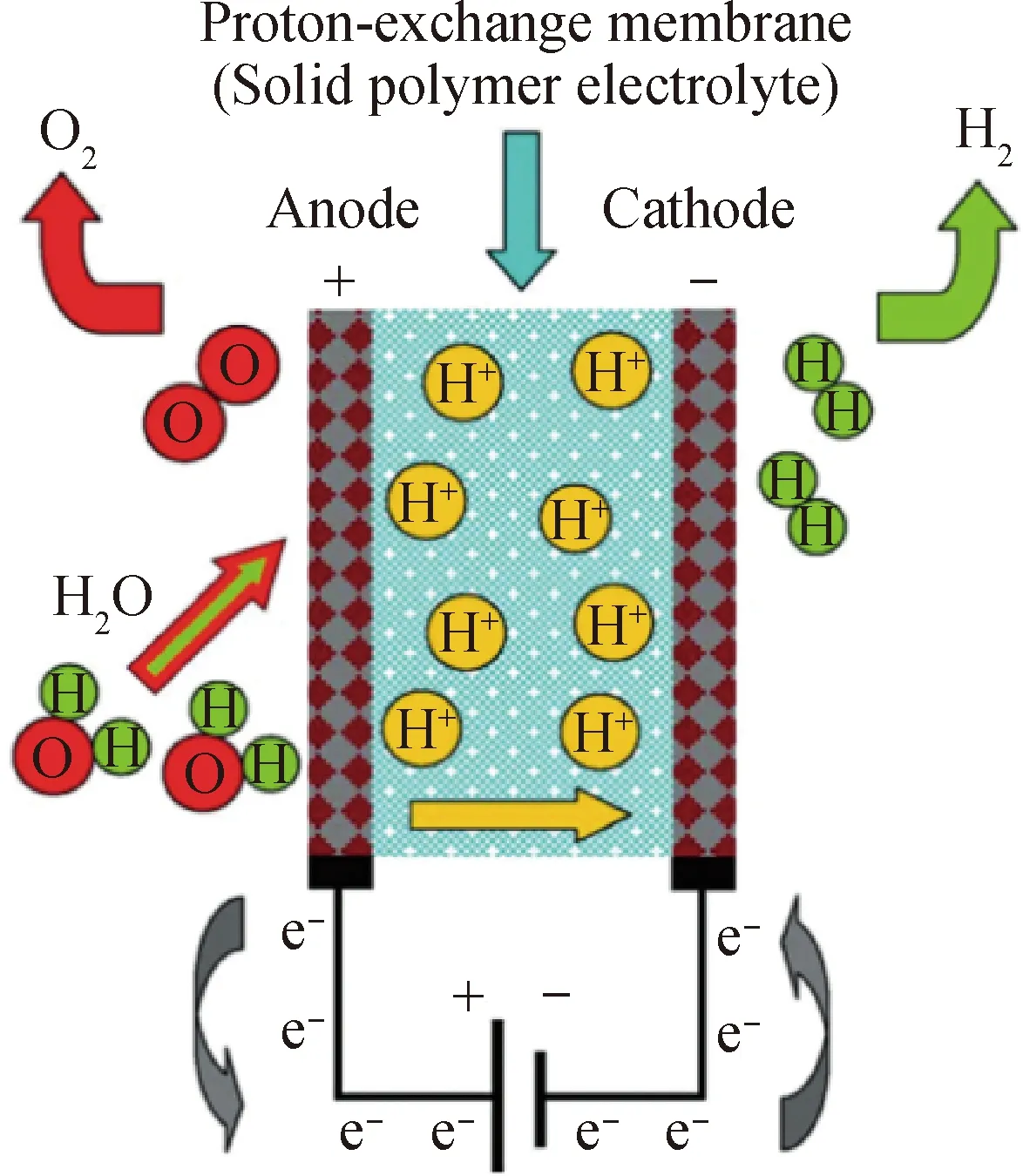
图2 质子交换膜水电解制氢原理[35]Fig.2 Principle of proton-exchange membrane water electrolysis for hydrogen production[35]
M+H2O→M-OHads+H++e-
(4)
M-OHads+M-OHads→M-Oads+M+H2O
(5)
or M-OHads→M-Oads+H++e-
(6)
M-Oads+M-Oads→2M+O2
(7)
or M-Oads+H2O→M-OOHads+H++e-
(8)
M-OOHads→M+O2+H++e-
(9)
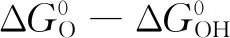
气相色谱条件:HP-5毛细管柱(30 m×0.32 mm×0.5 μm),程序升温,起始温度35 ℃,保持5 min,以4℃/min升至160℃,保持2 min,再以8℃/min升至220℃,保持3 min,进样口温度250℃。质谱条件:MS,电离方式为EI,电子能量70 eV,离子源温度230℃,成分组分通过NIST98图谱库检索、分析,再结合文献进行人工谱图解析确认检测物成分[5]。
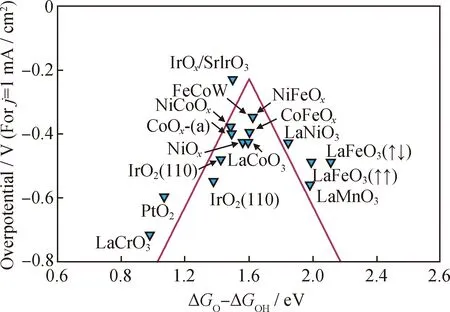
图3 金属氧化物的OER火山型图[38]Fig.3 OER volcano plot of metal oxides[38]
2 核-壳型Ir基催化剂的制备方法
催化剂的状态和微观形貌(包括催化剂的尺寸、结晶度、分散度、比表面积)对催化活性和稳定性有很大的影响,不同的制备方法对其形貌有很大的影响[32],所以选择合适的催化剂制备方法极其重要。
鉴于核-壳结构的特殊性,其制备方法也是特殊的,通常第一步合成核;第二步在核上形成壳。目前主要应用的核-壳结构催化剂的制备方法有胶体法(晶种生长法)、一步合成法、选择性脱合金法等。表1中列出了由不同制备方法制备的核-壳结构Ir基催化剂在酸性条件下的OER活性。
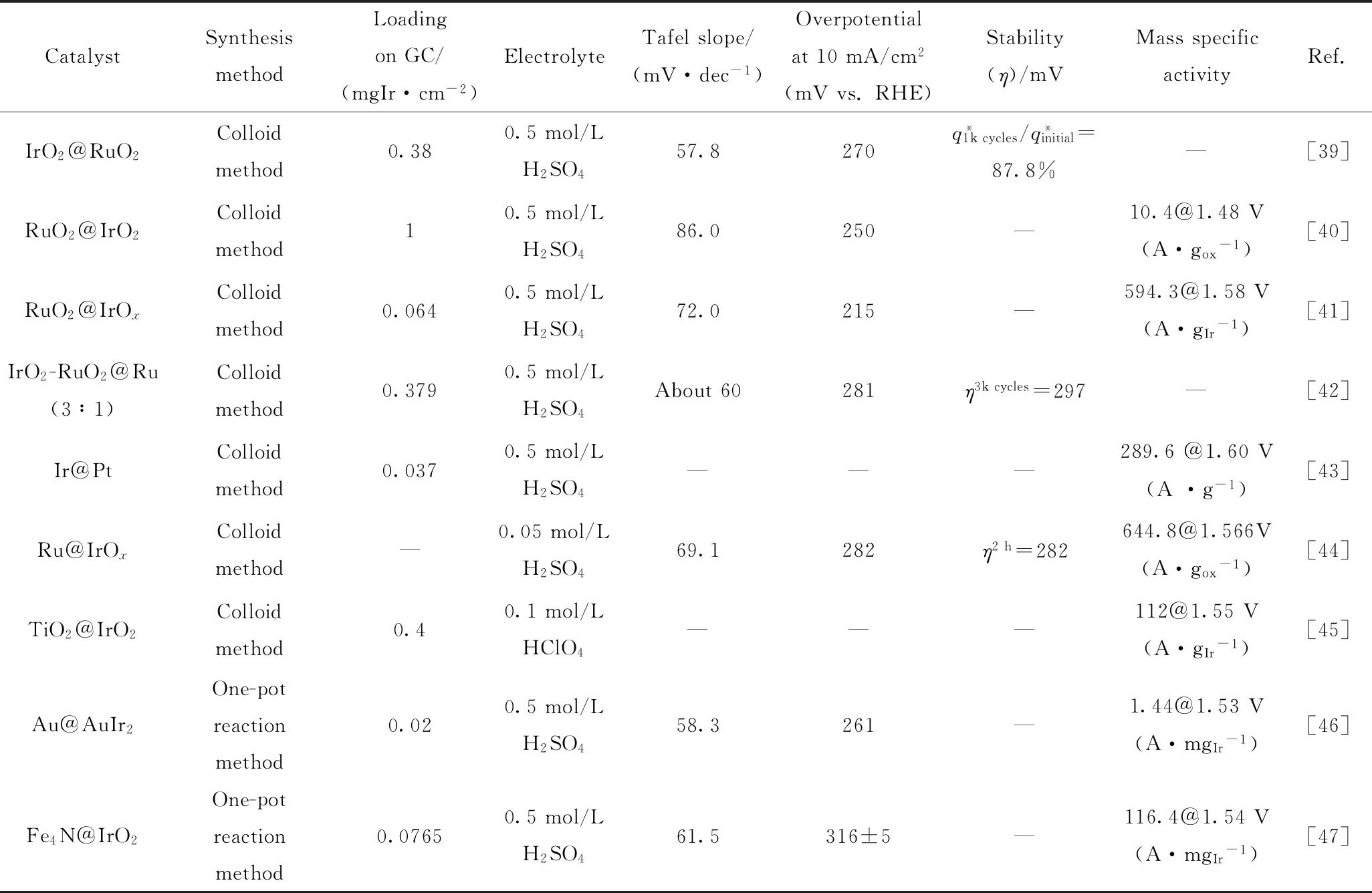
表1 不同核-壳结构Ir基催化剂在酸性条件下的OER过电势和质量活性的比较Table 1 Comparison of OER overpotential and mass activity of catalysts with different core-shell structures under acidic conditions
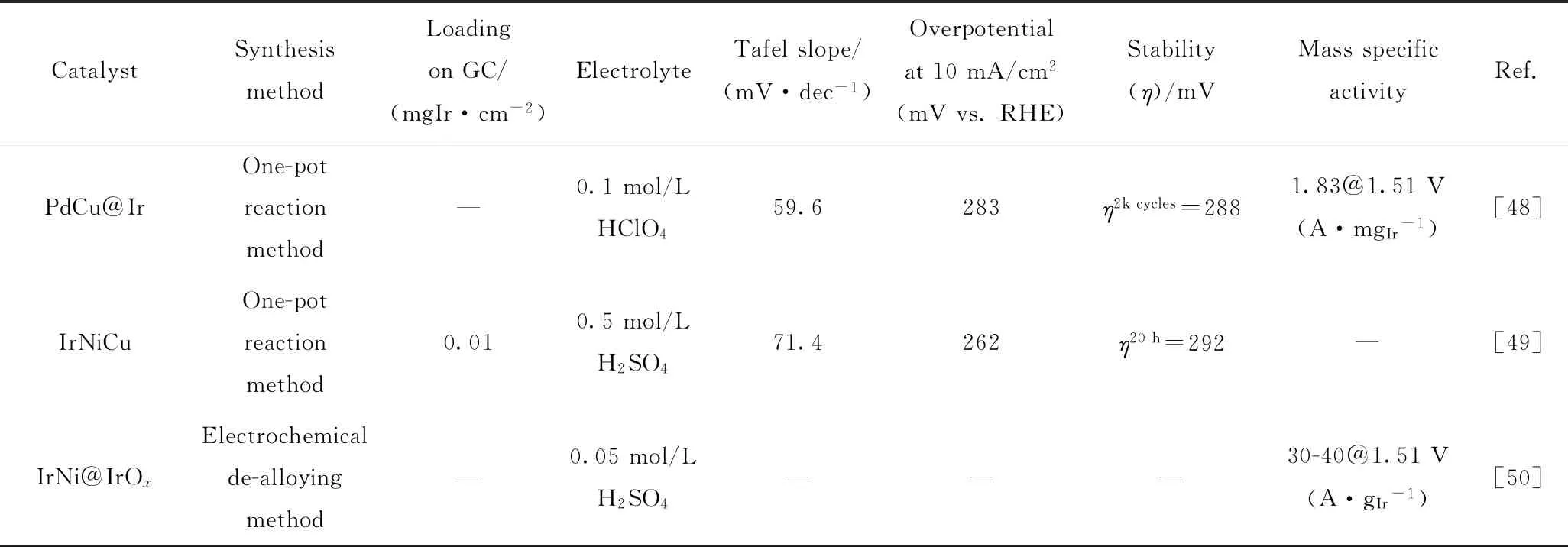
续表
2.1 胶体法(晶种生长法)
胶体法是合成核-壳结构催化剂最常用的方法之一。所谓胶体法,即首先在液相中制备出非Ir的胶体粒子,比如Cu、Ni等非贵金属作为胶体粒子,以此作为核和生长中心,或者使用已合成的非Ir粒子和溶剂混合作为核和生长中心,在此基础上还原Ir使之沉积在核上形成壳,通过控制加入前驱体的比例可以控制壳层的厚度,制备出活性和稳定性俱佳的核-壳型OER催化剂[51]。
Audichon等[39]研究了以商业RuO2为核和生长中心,在乙醇介质中还原IrCl3,制备出了IrO2@RuO2核-壳结构催化剂用于OER,通过透射电镜(TEM)、X射线能谱(EDS)证实了IrO2主要存在于催化剂表面,很好地包裹住了RuO2,并与商业RuO2和纯IrO2做了对比,结果表明,IrO2@RuO2具有最多的活性中心以及最大的可及性,且具有最高的稳定性,在电流密度为10 mA/cm2时过电势为270 mV,Tafel斜率为57.8 mV/dec,在0.3~1.2 V vs.RHE电位下10000次的循环伏安循环后保留了96.7%的初始电荷。研究显示,IrO2@RuO2的活性及稳定性的提高归因于2种金属元素的紧密接触,增强了IrO2和RuO2之间的协同作用。Ma等[40]以Ru/C为核,同样利用无水乙醇还原合成了RuO2@IrO2核-壳异质结构OER催化剂,获得了良好的活性,在1.48 V时,RuO2@IrO2的OER比活性是IrO2的3倍;该文献还着重阐明了大多数活性金属氧化物在酸性条件下的OER的反应机理:O—O键是通过在单个金属位置上的顺序水解离吸附而形成的;同时实验研究发现TiO2比RuO2更适合作为核,因为TiO2比IrO2的晶格常数大,而RuO2比IrO2的晶格常数小,通过晶格失配引起的晶格膨胀有利于促进IrO2上的OER,这一发现有助于为后续开展核-壳结构型催化剂研究提供理论依据。孙良良等[52]以NaBH4为还原剂,IrO2和Ti粉为主要原料,制备出了IrO2@Ti的核-壳结构催化剂,通过X射线衍射(XRD)分析发现IrO2@Ti中Ti的衍射峰很弱,说明IrO2已经成功包裹Ti形成了良好的具有核-壳结构的催化剂物质结构。
Lü等[41]利用乙醇还原法合成了RuO2@IrOx核-壳异质结构OER催化剂。X射线光电子能谱技术(XPS)分析表明,RuO2@IrOx纳米复合材料比其他样品具有更高的比活性、更大的质量活性、更小的过电位和更低的电池电位,这归因于核-壳异质结构处存在较高的电化学活性表面积(ECSA)和由协同作用引起的较快的动力学。在稳定性测试过程中,RuO2@IrOx阳极在300 h的稳定性实验中,在1 A/cm2时表现出良好的稳定性,这归因于非晶态的IrOx壳对于RuO2核的良好保护。
Li等[42]经过两步还原制备了间断覆盖的IrO2-RuO2@Ru(Ir/Ru摩尔比为3∶1)结构,其比表面积高、电荷阻力小、颗粒分布均匀的特征使得其在电流密度为10 mA/cm2时过电势仅为281 mV。Zhang等[43]在十六烷基三甲基氯化铵的存在下用NaBH4还原H2IrCl6,首先得到Ir纳米粒子,再加入H2PtCl6和还原剂抗坏血酸,使得还原出的金属Pt沉积在Ir纳米颗粒上形成Ir@Pt核-壳结构,其具有分散性好、分布均匀的特点,不仅对OER的催化活性高,而且对氧还原反应(ORR)也具有较高活性,Pt与Ir的相互作用和特殊的核-壳结构使得ORR和OER活性提高,这种新型的核-壳结构为开发高活性的双功能催化剂提供了方向,具有潜在的应用前景。Shan等[44]以聚乙烯吡咯烷酮为稳定剂、用乙二醇还原Ru(acac)3首先制备出了Ru胶体粒子,再高温还原IrCl3合成了Ru@IrOx核-壳结构OER催化剂。利用高度应变和无序的Ru核和部分氧化的Ir壳间的电荷重分布,使得Ir壳层价态的增加和Ru核价态的降低,激活了Ru和Ir之间的电子协同和结构相互作用,从而提高了催化剂的活性和稳定性。一方面,与RuIrOx相比,Ru@IrOx由于核-壳界面电荷重新分布使得Ru价态降低,Ir价态升高态,导致氧中间体的最佳吸附,促使其获得良好的催化性能;另一方面,由于核-壳结构对内在核金属有良好的保护作用,降低了金属的溶解,使得稳定性大大提高。通过合理促进异质结构内的电荷重新分布,实现催化剂活性和稳定性的同时,提高其催化性能是开发高效的电催化剂的一条有效途径。
Phama等[45]首先通过调节TiO2表面的正负电荷使得在其表面形成H2IrCl6壳,再通过热分解形成IrO2壳,获得TiO2@IrO2核-壳结构催化剂,提高了IrO2的分散性,该研究直接将催化剂沉积在多孔传输电极的传输层上,使得催化剂和多孔传输层集电器实现最佳接触,进而实现了活性金属Ir低负载量下的高活性,但是还需要继续优化热处理条件来实现高稳定性。
综上所述,胶体法是经过连续两步还原或者一步还原制得核-壳结构催化剂粒子,优点是可以制备出粒径较小、结构和尺寸可调控的纳米粒子,并且制备的装置较简单,但是存在稳定剂不易除去的缺点。
2.2 一步合成法
Wang等[46]以HAuCl4和IrCl3为原料、油胺为还原剂首次通过一步合成法合成了Au@AuIr2核-壳结构纳米颗粒催化剂,其对OER的本征活性和质量活性分别是商用Ir催化剂的4.6和5.6倍,并且其对氢析出反应(HER)的催化活性可以和Pt/C相媲美。X射线吸收精细结构谱(XAFS)和XPS的结果均表明,Au和Ir之间存在较强的电子相互作用,从而形成了部分氧化表面,平衡了中间产物和催化剂的结合力,使得OER催化活性大大增强。其OER稳定性也大大提高,是已报道的酸性介质中稳定性最好的催化剂。当Au@AuIr2同时作为阳极和阴极催化剂、电解槽的总电流为10 mA/cm2时,电解槽电压为1.55 V,并保持了40 h以上的活性,大大超过了商用阳极和阴极催化剂组合(Ir/C‖Pt/C,1.63 V,活性在几分钟内下降),是目前已报道的最有效的双功能催化剂之一。Liu等[54]以Ir(acac)3和IrCl3同时作为Ir源,Ni(acac)2作为Ni源,油胺为溶剂,以1,2-十六烷二醇为还原剂,利用其不同的反应活性,采用一步合成法合成了一种新型高效的IrNi@Ir核-壳催化剂,如图4所示。此方案的关键之处在于Ir(acac)3和IrCl3同时作为前驱体。研究发现,还原剂1,2-十六烷二醇的用量和纳米粒子的粒径密切相关,随着还原剂用量的增加,粒径减小。Tackett等[47]提出了用过渡金属氮化物作为核、Ir作为壳的核-壳结构型用于电解水OER的催化剂,研究了第VIII族过渡金属(Fe、Co和Ni)氮化物作为核材料,发现其中Fe4N@IrO2核-壳型纳米粒子表现出较强的OER活性和稳定性。Fe4N@IrO2的Ir/Fe摩尔比为1∶3,与纯IrO2相比,Fe4N@IrO2中Ir的质量降低了约50%,在酸性强氧化性的OER条件下,IrO2壳层对氮化物核起到保护作用,并且Fe4N@IrO2中单位质量的Ir比IrO2活性更高,稳定性更好。这一研究结果表明,Fe4N@IrO2成为降低Ir用量的有前景的候选方案。

图4 IrNi@Ir核-壳纳米粒子合成方案示意图[54]Fig.4 Schematic diagram for the synthesis of IrNi@Ir core-shell nanoparticles[54]
除了二元核-壳结构催化剂之外,一步合成法合成的三元核-壳结构催化剂也表现出优良的OER活性和稳定性。Li等[48]开发出了PdCu@Ir三元核-壳催化剂,具有优良的OER活性,而且通过合成相对较厚的Ir壳层消除了配体效应之后,研究了应变效应在提高酸性OER活性中的单独作用。密度泛函理论计算表明,Ir壳层上的压缩应变效应降低了含氧中间体的吸附强度,使d带中心下移,从而促进了氧气分子的生成。
Zhang等[49]利用一步合成法合成了具有富Cu@富Ni@富Ir多元合金和双核-壳结构,即最内层为Cu核,中间包裹Ni,最外层包裹Ir,如图5所示。由图5可以看出,Cu主要集中在整个催化剂结构的核心中间,表明在合成过程中Cu作为整个结构的中心优先形成核,然后在富Cu相上镀Ni,形成一定的取向,最后,Ir生长在富Cu@富Ni的3D多面体的最外层。特殊核-壳结构带来的较大的电化学活性面积,以及Ir、Ni、Cu 3种元素和双核壳结构的协同效应,使得IrNiCu不仅是优良的OER催化剂,而且对HER有很高的活性。IrNiCu双核-壳结构催化剂对OER表现出最高的电催化活性,在电流密度达到10 mA/cm2时,其过电位仅为262 mV,Tafel斜率为71.4 mV/dec,优于商业IrO2(326 mV/dec和77.3 mV/dec),并且稳定性良好,展示了一种新型的设计Ir基多金属双核-壳结构作为双功能催化剂的策略。
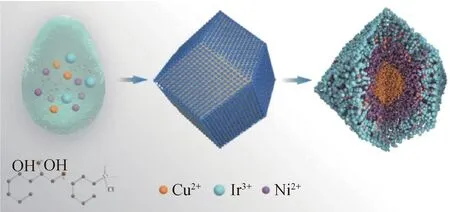
图5 IrNiCu双层核-壳结构的合成示意图[49]Fig.5 Schematic diagram of the synthesis of double-layer core and shell structure of IrNiCu[49]
一步合成法受限制于金属还原的电位,需要选择合适的前驱体,有些金属不适合该方法。
2.3 选择性脱合金法
选择性脱合金法的制备思路不同于寻常的核-壳结构制备方法。首先合成IrM(M为非贵金属)合金,然后通过电化学选择性脱合金或者酸浸蚀法将表面的非贵金属去除,可得到表面富集Ir的催化剂,再进行表面氧化,进而形成IrM@IrOx核-壳结构催化剂[53],如图6所示。
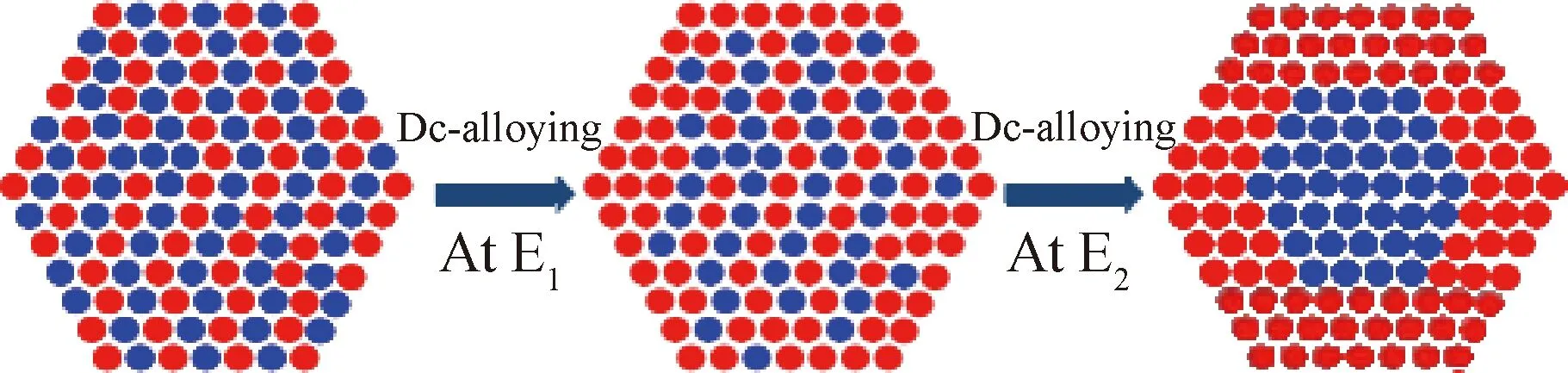
图6 电化学脱合金法制备核-壳结构催化剂[53]Fig.6 Preparation of core-shell structured catalyst by electrochemical de-alloying method[53]
Kim等[55]通过制备一系列IrxOs(1-x)合金,以此为前驱体,利用电化学快速脱合金法脱除Os,制备了金属Ir核-氧化物IrOx壳的高导电性纳米粒子,该结构平衡了OER催化剂的活性和稳定性,提出了准确衡量OER催化剂活性-稳定性因素(Activity-Stability factor (ASF)),证明了在相同过电势下,性能好的OER催化剂材料应该具备高的ASF值。其中以Ir25Os75为前驱体制备的脱合金核-壳结构催化剂的ASF是传统IrO2催化剂的30倍,这得益于金属Ir核-氧化物IrOx特殊结构的形成,而不是由于Os在Ir表面原子上诱导的电子效应所导致的。
Nong等[50,56]在以1,2-十四烷二醇作为还原剂、油酸和油胺作为封端剂的情况下,使用多元醇方法[57]制备出许多不同比例的负载在介孔锑掺杂的氧化锡(IrNix/Meso-ATO)上的IrNix(x=3.3~3.8)纳米粒子合金作为前驱体,通过电化学选择性脱合金以及选择性表面氧化制备了IrNi核-IrOx壳的催化剂。首次提出了低Ir含量的核-壳氧化物催化剂与高导电性、耐腐蚀氧化物载体的结合,解决了与贵金属含量和电解水阳极催化剂的耐久性有关的问题。在该研究中,考察了不同退火温度(180、250、300、400、500 ℃)对前驱体形态的影响,得出只有在不高于300 ℃的退火温度下处理的前驱体有理想的IrNi金属合金,且180 ℃为适合于脱合金及核-壳结构形成的最佳退火温度。核-壳的形态取决于粒子的大小,Ni从大颗粒的核心中浸出,形成空心的核-壳结构,而小颗粒则形成富Ni核的核-壳颗粒,如图7所示。贵金属的表面扩散速率必须小于非贵金属的表面溶解速率,才能使脱合金化深入到本体深处,从而产生纳米孔隙度。在过电势为280 mV下,IrNiOx/Meso-ATO-180的OER质量活性是IrOx/C和IrOx/COM-ATO的2.5倍,且在20 h的稳定性测试中电极电位几乎保持不变,耐腐蚀性载体为催化剂的稳定性起到了极大的促进作用。
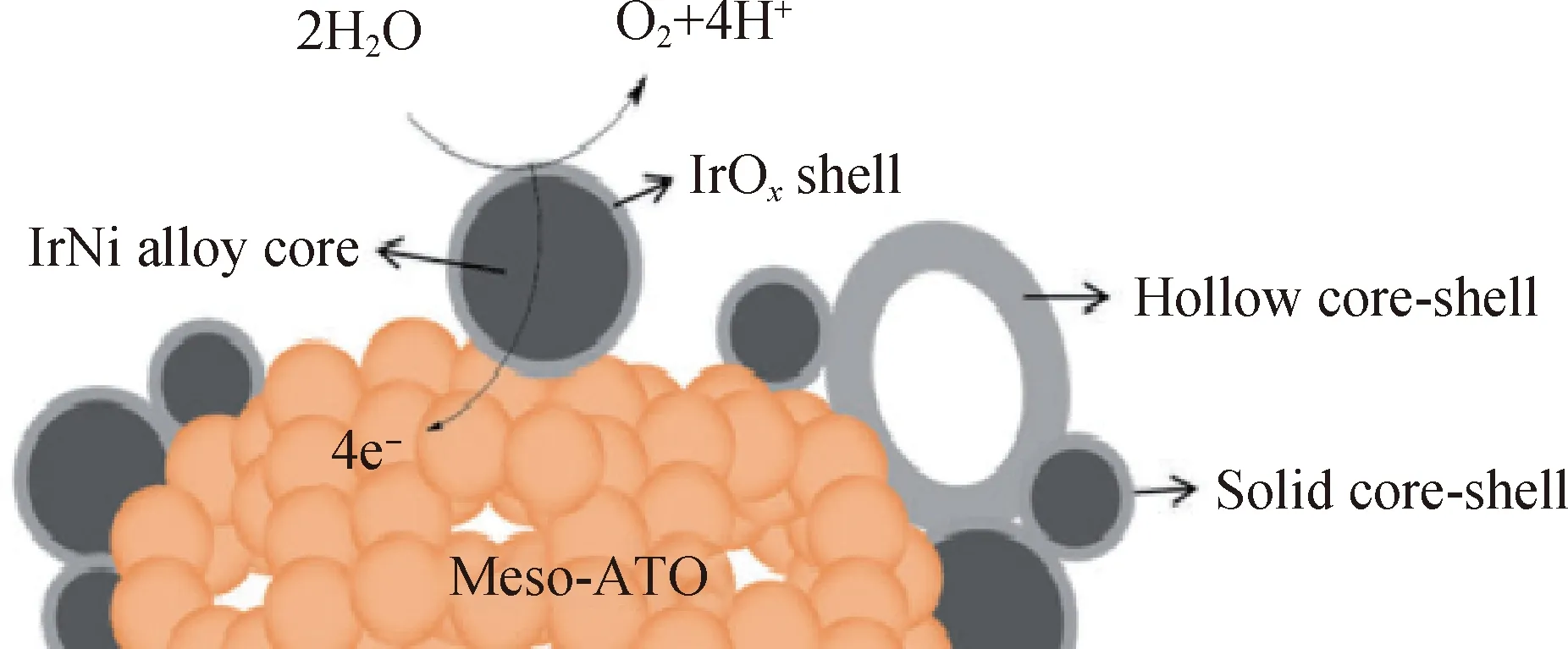
图7 负载在Meso-ATO上的核-壳型IrNiOx催化剂表面IrOx壳上的析氧反应方案[56]Fig.7 Scheme of oxygen evolution reaction on the surface IrOx shell of the IrNiOx core-shell catalyst on Meso-ATO[56]
3 核-壳结构型催化剂表征方法
研究人员在探究Ir基核-壳结构型催化剂制备方法方面做了深入研究,以探究最佳的设计方案,同时另一个不容忽视的环节是如何判断合成的催化剂是否具有核-壳结构[58]。Ir基催化剂的表征手段和其他催化剂类似,可通过TEM表征分布情况、粒径尺寸和形貌等,通过XPS分析催化剂表面的元素组成、相对含量和元素价态等,通过XRD确定晶体结构等,但是核-壳结构的存在仅通过TEM、XPS、XRD等表征无法来证明,需要更直观的表征手段来提供更有力的证据。在核-壳结构催化剂的表征过程中有其特有的表征手段,但多为几种方法联合来证明核-壳结构的存在,主要有高角度环形暗场(HAADF)-扫描透射电子显微镜(STEM)、能量色散X射线谱(EDX)、电子能量损失谱(EELS)等,为核-壳结构的构建提供有价值的信息[53]。
3.1 HAADF-STEM
STEM是结合扫描电子显微镜和透射电子显微镜2种电镜特征的具有更高分辨率的分析电子显微镜,是近几年发展起来的一种新型分析技术。STEM通常和HAADF联合使用可以获得暗场图像,因为HAADF的强度正比于元素原子序数的平方,所以不同原子序数的元素显示出不同的亮度,可以利用该特征来分辨核-壳结构,如图8所示[44]。由图8可知,Ir(77)和Ru(44)的原子序数相差很大,可以清楚地识别出具有较亮对比度的含Ir壳层和具有较暗对比度的Ru核。图9为Au@AuIr2核-壳粒子HAADF-STEM像和相应的EDX图像[46]。在图9 (a)中可以看到中心较亮、边缘较暗的图像,结合图9(b)~(d)相应的EDX面扫描图可以更加准确地判断出Ir主要分布在边缘处,而Au在核心处含量最高。
图8 Ru@IrOx纳米晶体的HAADF-STEM图[44]Fig.8 HAADF-STEM images of Ru@IrOx nanocrystals[44](a)HAADF-STEM image of a randomly chosen Ru@IrOx core-shell icosahedral nanocrystal along the [112] axis;(b)Simulated HRSTEM image of a Ru147@Ir414 icosahedral model structure along its [112] axis

图9 Au@AuIr2核-壳粒子HAADF-STEM像和相应的EDX图像[46]Fig.9 HAADF-STEM images and corresponding EDX images of Au@AuIr2 core-shell particles[46](a)HAADF-STEM image of Au@AuIr2;(b)Merged signal;(c)Au(gold);(d)Ir(blue)
3.2 EDX
利用EDX线扫分析材料成分时可以得到比较直观的结果。利用不同元素的特征X射线的波长、强度不同来检测元素[46],如图10所示。从图10可以观察到,在中心核位置处Ir的强度基本保持不变,且低于Au的强度,这表明形成了核心为纯Au金属、外壳为Au-Ir合金的Au@AuIr2核-壳结构。
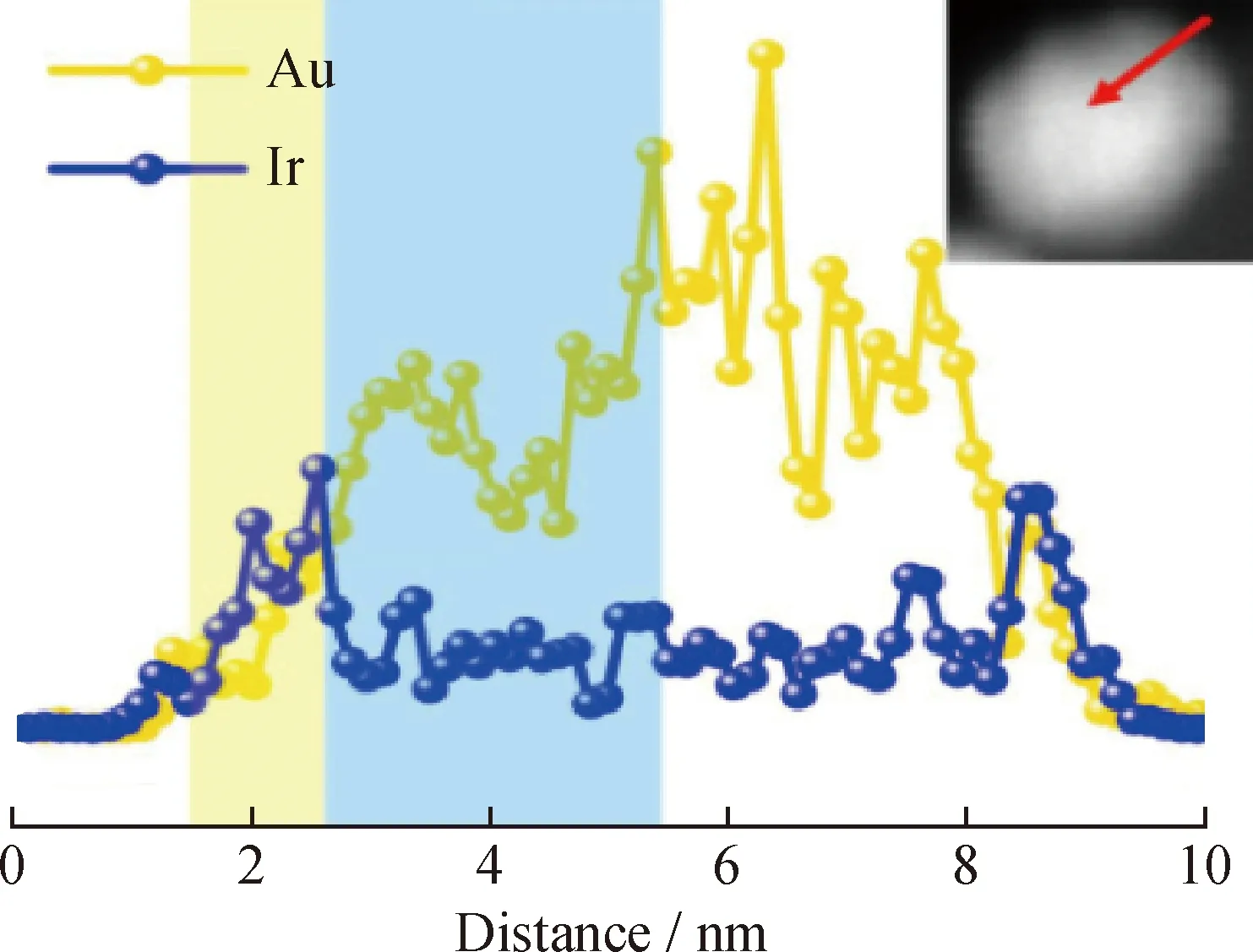
图10 沿着插图中标记的红色箭头对Au@AuIr2进行EDX线扫描分析[46]Fig.10 EDX line scanning analysis of Au@AuIr2 along the red arrow marked in the figure[46]
3.3 EELS
EELS和特征X射线都能表征物质的属性,但是EELS还能够提供化学键态、原子配位等结构细节。图11中显示了横跨球状Ru@Pt纳米粒子的横向扫描中的强度分布[59],其中Ru的EELS信号指示5.7 nm的纳米粒子内有4.2 nm的Ru核。

图11 有序Ru@Pt NP的HAADF STEM和Ru的EELS线扫描轮廓[59]Fig.11 EELS line scanning profile of HAADF STEM and Ru for an ordered Ru@Pt NP[59]
相比于一般纳米材料的表征来说,核-壳结构纳米粒子的表征更加复杂,判断是否为核-壳结构、确定核-壳结构壳层的厚度等等通常需要几种表征技术手段互相配合,综合分析,共同证明核-壳结构的存在以及其他详细结构信息。
4 结 语
提高贵金属Ir的利用率,开发高性能低Ir用量电催化剂对推动PEM电解水制氢技术的商业化具有重要意义。核-壳结构Ir基催化剂是能够实现低Ir催化剂的最有效的方法之一,笔者综述了用于PEM电解水制氢阳极析氧反应的Ir基核-壳结构型催化剂的研究进展,系统讨论了核-壳结构催化剂的制备方法及常用表征方法。目前核-壳结构催化剂的发展从制备方法、表征手段到形成机理仍存在着一些问题,其商业化应用尚有许多难题需要解决,笔者认为未来核-壳结构电催化剂的研究应主要集中在以下4方面:(1)探究核-壳结构合成过程中的内在相互作用机理,为高活性催化剂的可控制备提供理论依据。(2)设计可重复性的制备方法。寻找一种既可以控制核-壳结构催化剂的精细结构,又可以降低成本的大规模生产方法。(3)核心金属的晶格参数和壳层金属的匹配程度、核心金属的成本、核心金属前驱体的选择都会影响核-壳结构催化剂的结构的完整性,进而影响其活性和稳定性。(4)发展直观且精准的核-壳结构催化剂材料表征方法。目前表征核-壳结构催化剂的方法多为几种表征技术联用,表征过程繁多,有些技术表征结果不够直观、精准,应大力发展实时监测技术以及原位表征技术。
