改性渣油加氢催化剂硫化期活性相的研究
2021-12-22贾燕子徐广通
袁 蕙,贾燕子,徐广通
(中国石化 石油化工科学研究院 石油化工催化材料与反应工程国家重点实验室,北京 100083)
渣油固定床加氢处理是劣质重质油轻质化的最佳加工手段之一,能处理杂质含量高的渣油,不仅为重油催化裂化提供优质的原料,同时也减缓了下游装置设备的腐蚀。但渣油加氢催化剂约占生产成本费用的14%,因此催化剂使用寿命是制约装置长运转周期和控制加工成本的瓶颈[1-2]。
渣油加氢催化剂载体为氧化铝或二氧化硅等,而负载的活性金属主要为Mo等,助剂主要为Ni或Co,同时存在未助剂化的MoS2相、助剂化的加氢脱硫中心Ni(Co)MoS相和载体的酸中心[3]。固定床渣油催化剂装填后要经过高压硫化、VGO过渡、切渣后进行渣油加氢反应。催化剂由氧化态逐步转变为硫化态的过程,可称为硫化期。目前认为减缓初期催化剂活性相失活速率,能够增加其末期提温空间,有效延长渣油加氢装置运转周期,继而产生较好的经济效益。渣油加氢工业催化剂在硫化期活性相的结构与反应初期性能及失活速率紧密相关,但目前对不同阶段催化剂的活性相结构认识不足,沉积物(积炭等)对催化剂活性相的影响也不甚清楚[4-5]。
笔者以不同改性处理的γ-Al2O3为载体制备的2种NiMoP/Al2O3催化剂为研究对象,选取工业硫化梯度升温过程的低温中温点和高温终温点分别进行小型装置硫化,采用溶剂保护转移制样,系统原位表征其活性相,进而深入研究渣油加氢催化剂硫化期不同阶段活性相变化及沉积物对其的影响,将有助于分子尺度加深理解活性相的本质,控制积炭的形成、延长催化剂的使用周期,为催化剂配方和工艺条件的优化提供支撑。
1 实验部分
1.1 原料和试剂
硫化油,含二甲基二硫质量分数为2.5%的荆门常一线油,实验室自制。过渡油为茂名减压柴油(VGO),实验室自制。减压渣油A由中国石化仪长原油管道公司提供,其主要性质见表1。三氧化钼(MoO3),工业纯,天津四方公司产品;碱式碳酸镍(NiCO3·3Ni(OH)2·xH2O),工业纯,兴中科技公司产品;磷酸(H3PO4),分析纯,天津化工厂产品;γ-Al2O3,工业纯,中国石化催化剂长岭分公司生产;环己烷,分析纯,北京化工厂生产。

表1 减压渣油A的主要性质Table 1 Main properties of vacuum residue A
1.2 催化剂的制备
以γ-Al2O3作为载体,分别进行水蒸气改性和酸碱改性处理后,采用孔饱和浸渍法制备了金属负载量相同的NiMoP/Al2O3催化剂,然后经120 ℃干燥3 h,400 ℃焙烧3 h后备用。以水蒸气改性和酸碱改性的γ-Al2O3为载体制备的NiMoP/Al2O3催化剂分别记为SR和MG。其中w(MoO3)=14.0%、w(NiO)=3.5%、w(P2O5)=3.0%。
1.3 催化剂的硫化及反应活性评价
由于高通量装置高压硫化过程中难以采样,因此2种NiMoP/Al2O3催化剂分别在自制小型装置采用硫化油进行硫化实验。2种催化剂SR和MG分别以升温速率30 ℃/h从室温升温至230 ℃时恒温4 h后取样,样品未完全硫化,分别记为SR-M和MG-M;以升温速率30 ℃/h从室温升温到370 ℃时恒温4 h后取样,样品已经完全硫化,分别记为SR-E和MG-E。取样后样品均快速转移到装有环己烷的样品瓶中,隔绝空气保存,待用。
催化剂的反应活性评价在德国HTE高通量反应装置上进行。在高压14 MPa下将2种渣油加氢催化剂分别进行分步升温硫化完全后,更换茂名减压柴油(VGO)进行过渡,然后以减压渣油A作为原料,在反应温度385 ℃、反应压力14 MPa、质量空速0.5 h-1、反应时间24 h和48 h的条件下进行渣油加氢反应。根据减压渣油原料中的硫质量分数(w0,%)和减压渣油产品中的硫质量分数(w1,%)计算加氢脱硫率(rHDS,%)评价指标,计算公式如式(1)所示。
(1)
反应时间为24 h时,在完全硫化的MG和SR催化剂作用下减压渣油A的加氢脱硫率分别为45%和41%;反应时间为48 h时,在完全硫化的MG和SR催化剂作用下减压渣油A的加氢脱硫率分别为41%和38%。这说明相同条件下,MG加氢脱硫活性略高于SR。
1.4 催化剂的表征
(1)催化剂热解性质分析:采用德国耐驰公司生产的STA409PC-QMS403热重-质谱联用系统(TG-MS),扫描模式为MID模式,吹扫气为Air,流量为80 mL/min;保护气为N2,流量为20 mL/min;温度范围为35~800 ℃,升温速率为5 ℃/min。
(2)催化剂活性相微观形貌的HRTEM表征:采用美国FEI公司生产的Tecnai G2 F20 S-TWIN高分辨透射电子显微镜(HRTEM)观察活性相的形貌变化。收集少量的样品悬浮液于微栅上,每个样品采用相同的放大倍数和加速电压(200 kV)随机拍摄约20张照片,人工进行条纹相长度、层数和个数的统计[6]。
(3)催化剂积炭和活性相的Raman表征:采用日本HORIBA公司生产的LabRAM HR UV-NIR型激光共聚焦拉曼光谱仪(Raman),激发光源波长325 nm,紫外15倍物镜,共焦针孔直径100 μm,扫描2次取平均值。
(4)催化剂活性相的XPS表征:采用美国Thermo Scientific公司生产的ESCALAB 250型X射线光电子能谱仪,激发源为单色化AlKα X射线,功率为150 W。窄扫描所用通透能为30 eV。结合能用污染碳的C 1s峰(284.8 eV)校正。样品原位制样并转移后,抽真空12 h后移入样品室进行多元素的采谱和科学拟合。
(5)催化剂活性相的CO原位红外表征:采用美国Thermo Fisher公司生产的Nicolet 6700型傅里叶变换红外光谱仪,分辨率4 cm-1,扫描64次。原位转移的样品在有机溶剂保护下快速压制成直径为13 mm的自支撑片,放入自制石英红外样品池,进行150 ℃真空脱气净化4 h,再液氮降温至-190 ℃,饱和吸附已净化的CO探针分子0.5 h后进行脱附。将吸附前后的谱图进行差减,得到样品吸附CO的红外光谱图,并进行样品质量校正。
2 结果与讨论
2.1 催化剂热解性质的TG-MS表征
采用热重-质谱联用技术(TG-MS)全面表征不同硫化状态催化剂在空气中的热解行为。不仅可以通过热重曲线反映不同温度下各物种氧化燃烧的难易程度,而且可通过质谱分析来获取释放出的H2O、CO2和SO2等成分的定性和定量信息,来深入研究不同硫化期催化剂的含氢、含碳和含硫物种的组成和关联,从而推断硫化过程活性相的形成机理和积炭前驱体的形成和沉积状况[7-10]。
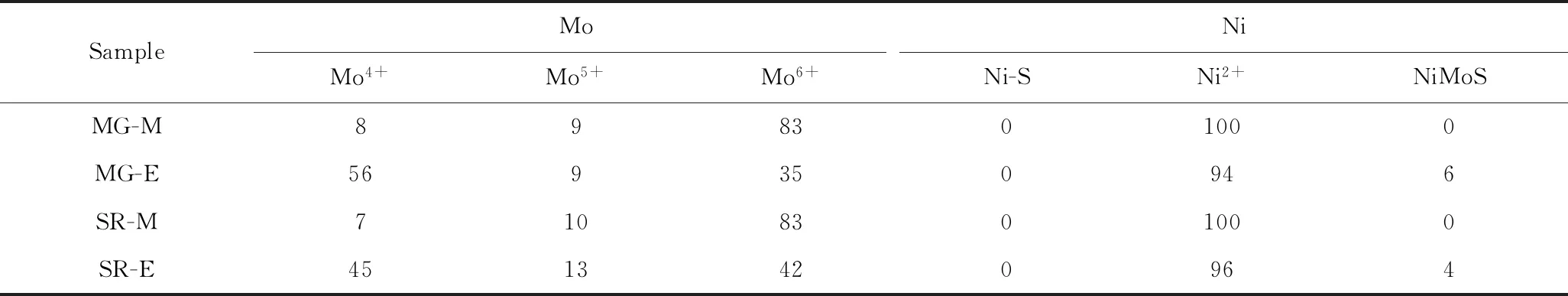
图1为MG、SR系列样品的TG-MS谱图,根据热重曲线统计的不同硫化条件制备的催化剂样品热分解过程各温度段质量损失率见表2。按照温度范围,样品的失重存在4个明显区域:第1区为T≤150 ℃;第2区为150 ℃ 图1 空气气氛下MG和SR催化剂样品的TG-MS谱图Fig.1 TG-MS spectra of MG and SR catalyst samples in air(a)MG-M;(b)SR-M;(c)MG-E;(d)SR-E 表2 MG和SR催化剂样品热分解过程各温度段质量损失率Table 2 Percentage of mass loss of MG and SR catalyst samples during temperature programmed oxidation w/% 为进一步研究硫化过程中各物种定性定量关系和形成过程,将SO2和CO2质谱曲线(图1)进行科学的分峰,分峰拟合定量结果如表3所示。由表3中SO2质谱曲线的分峰结果可知,MG-E和SR-E总体SO2质谱峰面积均约为MG-M和SR-M的2倍,且4个硫化物燃烧峰的峰温均比相应的MG-M 和SR-M的峰温高,说明随着硫化温度升高,硫化态活性相的数量大幅增加且活性相的类型向高温稳定的硫化态逐渐转变。SO2主要由硫化物的热解产生,根据对催化剂的认识及相关研究[7-10],推测催化剂在氧化燃烧过程中发生的主要反应列在表3中。MG-E和SR-E在290 ℃附近的峰面积最大,占总释放量的79%~82%,而在430、520和750 ℃附近还有3个较弱的谱峰,分别约占总释放量的10%、5%和1%,说明硫化态活性相大部分以MoS2形式存在,也存在少量NiS和硫酸盐形式,而随着温度升高,体相硫化态活性相占比有所提高。总体来说,MG系列和SR系列样品差异性变化规律一致,但和SR系列样品相比,MG系列样品各SO2燃烧峰对应温度略低,推测MG系列样品较易于硫化。 由表3中CO2质谱曲线的分峰结果可知,4种催化剂样品均有3个燃烧峰,但峰温稍有不同。第1个位于200~230 ℃,占总CO2释放量的10%~20%,出峰的温度较低,推断此处含碳化合物缩合度较低,可能与表面金属硫化物的初步分解有关,为活性相表面吸附的有机化合物。第2个峰位于280~320 ℃,约占总CO2释放量的10%,而该温度下SO2谱峰占总SO2面积比例很高(硫化完全样品达到约80%),且此处H2O燃烧峰面积占H2O总量也达40%,说明此时有大量硫化物的燃烧释放,而伴随燃烧的含碳氢化合物缩合度并不高。推测这是由于刚生成的硫化活性相活性较高,加氢能力强,且表面反应热较易扩散,因此不容易产生缩合度高的积炭。第3个峰位于380~470 ℃,所占比例达60%~90%,而此处H2O燃烧峰的占比仅为10%,SO2谱峰的占比也约为10%,说明此部分含碳化合物的缩合程度较高。推测可能为硫化过程中体相活性中心吸附和反应的碳氢化合物,虽然活性相加氢活性高,但是体相与表面相比,热扩散和物种扩散均较慢,因此易形成缩合度高的积炭前驱体物种。4种催化剂样品进行对比,发现低温硫化样品MG-M和SR-M中含硫物种比较分散,第3个CO2燃烧峰峰温更高且峰面积最小,推测可能为含钼的硫氧化物(MoOxSy)加氢活性较低,易积炭,因此导致其上积炭物种缩合度高[7]。但是继续高温硫化,随着硫化态物种的转变,此种积炭类型也发生变化。由此,TG-MS表征结果证实了在催化剂硫化过程中伴随着有机化合物的吸附、逐步缩合并逐步转化为积炭物种的过程。 表3 MG和SR催化剂样品TG-MS谱图的分峰拟合结果Table 3 Fitting results of TG-MS spectra for MG and SR catalyst samples 采用高分辨电镜(HRTEM)分析了MG和SR催化剂样品的活性组分的分布情况,典型的HRTEM照片见图2。依照已建立的方法分别对随机拍摄的催化剂电镜照片中MoS2片晶的特征进行人工统计[6]。由图2可以看出;MG-E和SR-E催化剂样品的活性相的平均长度分别为3.10 nm和3.05 nm,活性相的平均堆叠层数分别为1.70和1.78;低温硫化样品MG-M和SR-M均未见规则的MoS2条纹和堆垛,但分布着大量纳米黑色圆形聚集小簇,为MoS2条纹的前驱体。对比MG和SR催化剂,MG-M的前驱体纳米小簇比SR-M的小簇尺寸稍大、个数稍多,高温硫化MG-E样品的MoS2片晶比SR系列平均长度略长、层数略低。由此推断MG系列催化剂载体经酸碱改性使得载体与活性相作用较弱,更利于活性相的硫化和生长[6,11]。 图2 MG和SR催化剂样品的HRTEM照片Fig.2 TEM images of MG and SR catalyst samples(a)MG-M;(b)SR-M;(c)MG-E;(d)SR-E 表4 催化剂在氧化燃烧过程中可能发生的主要反应Table 4 Possible reactions during temperature programmed oxidation for catalyst samples X射线光电子能谱仪(XPS)是重要的表面分析技术手段之一,能够给出催化材料活性组分的类别、化学态、在载体表面的分散状态及其与载体的相互作用等。因此将SR和MG 2种催化剂硫化后进行原位制样密闭转移,再进行XPS表征和分峰拟合,获得硫化过程中催化剂表面活性组元的变化。不同硫化状态下的4种催化剂样品各元素与Al的摩尔比见表5。随着硫化深度增加,样品的n(S)/n(Al)和n(C)/n(Al)增大,说明活性金属硫化过程逐步深入,同时含碳化合物伴随着增加。n(Ni)/n(Al)没有变化,说明金属元素Ni的化合物一直分布在催化剂的表面。而n(Mo)/n(Al)和n(P)/n(Al)略有增大,可能元素Mo和P随着硫化进程会迁移到表面。 表5 MG和SR催化剂样品的XPS表征结果Table 5 Fitting results of MG and SR catalyst samples by XPS 加氢催化剂中的活性元素通常具有多种化学态[6,12],MG和SR催化剂样品中Mo、Ni不同化学态的摩尔分数见表6。由Mo 3d5/2结合能,可知渣油加氢催化剂的Mo有3种化学态:229.0 eV的峰对应于+4价的硫化态Mo,230.2 eV的峰对应于+5价的氧硫态Mo,232.6 eV的峰对应于+6价的氧化态Mo。由Ni 2p3/2结合能,可知Ni也存在3种化学态:硫化物中的Ni(用Ni-S表示,约853.1 eV)、NiMoS活性相中的Ni(约854.6 eV)、氧化态Ni(用Ni2+表示,约856.4 eV)。由Mo4+占总Mo的摩尔分数来代表硫化度,从表5可知低温硫化样品(MG-M 和SR-M)的硫化度仅为7%~8%,高温硫化样品(MG-E和SR-E)的硫化度能达到45%~56%。但并未看到助剂金属Ni硫化聚集态的存在,说明元素Ni均匀分散在催化剂上。硫化态NiMoS活性相在MG-E中摩尔分数为6%,略高于SR-E中的4%。与CO原位红外表征结果相一致。载体酸碱改性处理的催化剂MG系列减弱了金属与载体相互作用,会促进活性金属在载体表面分散且硫化度有所提高。 表6 MG和SR催化剂样品中Mo、Ni不同化学态摩尔分数Table 6 The mole fraction of different chemical states of Mo and Ni in MG and SR catalyst samples x/% 图3 MG和SR催化剂样品的Raman谱图Fig.3 Raman spectra of MG and SR catalyst samples CO为弱碱性探针分子,通常利用红外光谱研究其在催化剂表面的吸附后特征峰位的变化来提供催化剂表面结构、活性中心方面的信息。针对硫化渣油加氢催化剂摸索建立了CO探针低温原位红外表征方法(CO-in situ IR)及合适的原位转移和制样方法,能够清晰地表征各活性相,结果见图4。由图4可以看出:4种催化剂样品均在2182、2154、2130~2000 cm-1处产生3个明显的CO特征吸收峰:2182 cm-1处特征峰归属于与Al2O3载体中的Al3+Lewis酸中心[15];2154 cm-1处特征峰归属于与Al2O3载体表面弱酸性OH中心氢键;2130~2000 cm-1处特征峰归属于金属硫化态活性相的CO吸附,为多个光谱特征峰的叠合。依据渣油加氢催化剂的特点进行科学归属,其中2130 cm-1处特征峰为助剂化NiMoS活性相中Ni中心的CO吸附[5,16],2110 cm-1处特征峰为无金属助剂作用的六配位Mo中心[6],2095 cm-1处特征峰可能为NiSx物种中Ni中心[15-16],2080 cm-1处特征峰归属为与Ni相邻的五配位Mo中心[4,16],2060 cm-1处特征峰归属为与Ni相邻的四配位Mo中心[4]。 由图4可知,高化学敏感性的CO探针给出了催化剂不同活性中心的分布,相比其他表征手段更加丰富。为进一步研究各样品活性中心的种类和相对大小,对谱图进行了分峰拟合。结果表明,4种催化剂样品从低温部分硫化到高温进一步硫化,波数2182 cm-1和2154 cm-1处载体2个特征峰的变化不大,但是硫化态活性相的CO特征峰面积约为低温硫化样品的2倍,其中2110 cm-1和2080 cm-1处活性相Mo中心增长倍数大于2130 cm-1和2095 cm-1处活性相Ni中心,由此推测Ni中心比Mo中心更容易硫化,高温硫化才能使Mo中心完全硫化。MG催化剂样品因为载体酸碱改性使得载体的Lewis酸中心数量和OH酸中心数量均比SR催化剂样品低,但其金属活性相峰面积比SR催化剂样品略大。由CO-NiMoS与CO-MoS2的积分面积之比(ACO-NiMoS/ACO-MoS2)可以给出硫化态催化剂的助剂化作用的程度的信息。经分峰拟合计算,MG-E的ACO-NiMoS)/ACO-MoS2的比值为0.86,略高于SR-E样品的0.80。说明MG-E的助剂化程度更高,可能归因于载体酸碱改性减弱了金属与载体相互作用,会促进活性中心类型有所变化。 图4 MG和SR催化剂样品的CO吸附红外光谱Fig.4 CO-in situ FTIR spectra of MG and SR catalyst samples 通过建立CO探针变温原位红外表征方法,可以更清晰展现出上述活性相的特征峰,并得到各活性中心和载体酸性中心的电子分布状况。MG-E催化剂样品的变温CO谱图见图5。由图5可见,2200~2135 cm-1范围内与载体作用的CO较容易脱附,而位于2135~2000 cm-1范围内与硫化相结合的CO较难脱附。这说明与载体吸附位相比,硫化相吸附中心吸附CO的能力和接收电子的能力更强。比较不同硫化相上CO的吸附峰强度随温度的变化,得出与MoS2中心结合的CO较易脱附,而与NiMoS中心(在2080 cm-1和2060 cm-1)作用的CO较难脱附。这一结果表明,NiMoS相比MoS2活性中心接受电子能力更强。对于加氢脱硫反应,由于渣油中含硫分子中硫原子的孤对电子与缺电子的NiMoS活性中心存在较强的σ配位键,键合作用较强,导致噻吩环中的C=S键容易断裂[4,16]。因此,NiMoS活性中心数目的增加对应高的HDS活性。MG-E的NiMoS活性中心比SR-E数量多,且助剂化作用略强,因此加氢脱硫能力略强。这与反应活性评价结果一致。 图5 升温脱附过程中MG-E样品的CO吸附红外光谱Fig.5 CO-in situ FTIR spectra of MG-E during temperature programmed desorption 研究含碳物种所采用的方法为TG-MS表征催化剂氧化燃烧过程(见2.1节),但渣油加氢催化剂硫化和反应过程处于无氧环境。为研究无氧环境下含碳物种随温度升高能否脱附和分解,设计氮气条件下对MG-E样品进行程序升温TG-MS表征,结果见图6。由图6可见,MG-E在氮气条件下升温至800 ℃约有8%的失重,主要为H2O,其中145 ℃主峰和250 ℃肩峰分别为表面和体相吸附水,而高于460 ℃时仅占总峰面积20%,可能为样品的结构水。质谱全谱扫描结果表明,氮气升温过程中MG-E样品没有明显的大分子有机物分子脱附,只有质荷比14的物种在240 ℃附近有1个很小脱附峰,可能为催化剂上吸附的小分子易挥发有机物。 图6 氮气气氛下MG-E样品的TG-MS谱图Fig.6 TG-MS spectra of MG-E catalysts in N2 依据工业催化剂渣油加氢反应温度为360~395 ℃以及TG-MS研究的结果,设计进行150、250、300和370 ℃下的真空净化过程来考察MG催化剂活性相的变化,采用低温CO探针原位红外表征各酸性中心和金属活性相,结果见图7。由图7可见,MG系列,随温度升高,吸附的CO特征峰总面积逐渐增大,即催化剂酸性中心和活性中心的数量逐渐增加。对比不同温度的吸附谱图,可知低温硫化样品MG-M的载体Al3+和OH酸中心数量随温度升高略微增大,金属活性中心稳步增长,但在370 ℃时,金属活性中心数量大幅增长,达到150 ℃时的4倍。MG-E的变化规律和MG-M基本相同,但其金属活性中心数量370 ℃时增幅比MG-M小,约为150 ℃时的2倍。进行谱图拟合分峰,计算得370 ℃下MG-E的ACO-NiMoS/ACO-MoS2比值达到0.94,高于为150 ℃时的0.86,说明活性金属助剂化程度增加,且强缺电子的低配位数的NiMoS相的比例增加。随净化温度升高,催化剂的酸性中心和活性中心表面附着的可挥发含碳物种可能逐渐被净化,使其暴露量增大,CO吸附量增大。而TG-MS结果表明,无氧净化条件下没有检测到明显的有机物脱附峰,因此活性相的增加可能主要归因于催化剂在高温条件下金属活性相的二次生成或重排。 图7 MG系列样品在不同净化温度下的CO吸附红外光谱Fig.7 CO-in situ FTIR spectra of MG-E and MG-M at different purification tempretures(a)MG-E samples;(b)MG-M samples 采用紫外拉曼光谱表征MG-E和SG-E样品和370 ℃净化后样品,对比考察金属氧化物、硫化物和积炭物种的变化,见图8。由图8可见,MG-E和SR-E位于1604 cm-1、1377 cm-1附近积炭的G峰、D峰在370 ℃净化后峰面积有所下降,D峰与G峰比值下降,说明净化过程主要除去了催化剂上的易挥发含碳物种,而部分聚合度高的积炭前驱体物种仍沉积在样品表面,与MG-E相比较,SR-E高温净化后的含碳物种较多,可能更易积炭。位于880 cm-1和948 cm-1处的较弱的拉曼特征峰消失,说明氧化态钼物种净化后消失,变成了硫化态钼物种。进一步证实了硫化态活性相在高温下可以发生动态变化,促使活性相的硫化度提高。370 ℃净化后MG-E和SG-E在381、409 cm-1处Mo-S的特征峰发生红移且半峰宽变大,说明净化后MoS2微晶的助剂化效果变强[13],即NiMoS含量占比更大。 图8 MG-E和SR-E升温净化前后的Raman谱图Fig.8 Raman spectra of MG-E and SR-E before and after purification 采用XPS表征MG-E硫化后样品及其370 ℃净化后样品,对比结果得到,高温净化后金属活性相的硫化度从56%提高到73%,硫酸盐占含硫物种摩尔分数由42%降为17%,NiMoS占总Ni物种摩尔分数由6%提高到45%。说明高温净化后部分硫酸盐变成了硫化态活性相,助剂化效果提升。这些均与原位红外结果一致。 MG-E、MG-M和SR-E、SR-M催化剂样品在370 ℃净化后进行低温CO探针原位红外表征,谱图见图9。由图9可见:和图7中其他温度谱图相比,370 ℃ MG-E和MG-M谱图特征峰的位置、比例几乎未变,仅大小发生变化,即各种活性相的比例未发生大的变化。说明370 ℃能够使得不同硫化程度的样品继续硫化直到完全。MG催化剂样品CO吸附峰面积大于对应SR催化剂样品,可见MG催化剂活性中心数量较多,其NiMoS占比稍高。这可能归因于载体改性使得载体与活性相作用减弱,硫化度提高,助剂化效果较好,加氢脱硫活性有所提升。 图9 MG和SR系列升温净化后的CO吸附红外光谱Fig.9 CO-in situ FTIR spectra of MG and SR series after purification 氮气TG-MS结果表明,300 ℃以上MG-E并没有明显的大分子有机物峰,说明聚合度高的积炭此时并没有离开体系,因此净化温度达370 ℃时金属活性中心数量大幅度提升,归因于高温硫化活性相的重排和二次生成。因此升温净化过程一定程度模拟了硫化过程的活性相动态转变过程,伴随着积炭前驱体物种的脱附和变化过程。而370 ℃净化后催化剂活性中心的特征更能体现渣油加氢反应中与渣油实际接触370 ℃反应的活性相的实际状态。催化剂初期失活是积炭和活性相的变化共同引起的,合适的酸中心和活性中心密度、合适的助剂化程度、能够在反应温度下保持活性相的平衡浓度和积炭前驱体脱附转化能力是理想的渣油加氢催化剂硫化后的特征。 (1)以渣油加氢NiMoP/Al2O3催化剂为对象,成功构建了TG-MS、TEM、XPS、Raman、CO探针低温原位红外等系统表征方法,能够全方位提供硫化期不同阶段样品的活性中心的类型分布、微观形貌、化学态和配位状态等分子水平的详细信息,使其有机结合并相互印证。 (2)研究硫化过程中改性渣油加氢催化剂活性相的形成和积炭前驱体的吸附转化过程。结果表明,随着硫化的深入,硫化度逐步增加,纳米黑色圆形小簇聚集形成条纹相,NiMoS相占比逐步提高,同时含碳物种的含量增加且聚合度增大。 (3)研究水热改性和酸碱改性载体制备的2个系列样品的活性相构效关系。结果表明,和SR催化剂样品相比,MG催化剂样品较易硫化,MoS2条纹较长且层数稍低,NiMoS相占比稍高,说明载体改性使得载体与活性相作用减弱,促进活性金属在载体表面分散,提高硫化度,进而提高加氢脱硫活性。虽然MG催化剂样品氧化还原温度较低较含碳物种较多,但高温净化后易去除。 (4)设计反应来探讨改性渣油加氢催化剂活性相的动态变化和沉积物的影响。升温净化后,部分聚合度较低的含碳物种被除去,金属氧化态物种和硫酸盐完全被硫化。与载体酸性中心相比,金属活性中心数量大幅增加,低波数的强缺电子NiMoS相比例显著提高。一定程度模拟了硫化过程中活性中心的动态重排、转变和新生过程,同时伴随着积炭前驱体的脱附和生成过程。催化剂初期失活是积炭和活性相的变化共同引起的,合适的酸中心和活性中心密度、合适的助剂化程度、能够在反应温度下保持活性相的平衡浓度以及积炭前驱体脱附转化能力是理想的渣油加氢催化剂硫化后的特征。


2.2 催化剂活性相微观形貌的TEM表征


2.3 催化剂活性相赋存状态的XPS表征

2.4 催化剂活性相的Raman表征


2.5 催化剂活性相的CO-原位红外表征


2.6 催化剂活性相动态变化和沉积物种的影响




3 结 论
